Whole-Genome Bisulfite Sequencing Data Standards and Processing Pipeline
Assay Overview
Whole Genome Bisulfite Sequencing is used to investigate DNA methylation patterns to base granularity. A bisulfite treatment converts cytosines into uracils, but leaves methylated cytosines unchanged. After mapping bisulfite sequencing reads against a Bismark-transformed genome, the pipeline extracts the CpG, CHG, and CHH methylation patterns genome-wide. CpG sites are palindromic, consisting of a cytosine nucleotide followed by a guanine, and are highly prone to methylation. Methylation increases the possibility of mutation in which cytosine is deaminated and becomes thymine. Methylation can also occur at CHG and CHH sites, where "H" is A, C, or T (Guo et al. Characterizing the strand-specific distribution of non-CpG methylation in human pluripotent cells, Nucleic Acids Res (2014) 42 (5): 3009-3016).
Updated June 2017
Pipeline Overview
The WGBS pipeline was developed as a part of the ENCODE Uniform Processing Pipelines series 1. The full WGBS pipeline code is available on Github and can be run on DNAnexus (link requires account creation) at their current pricing.
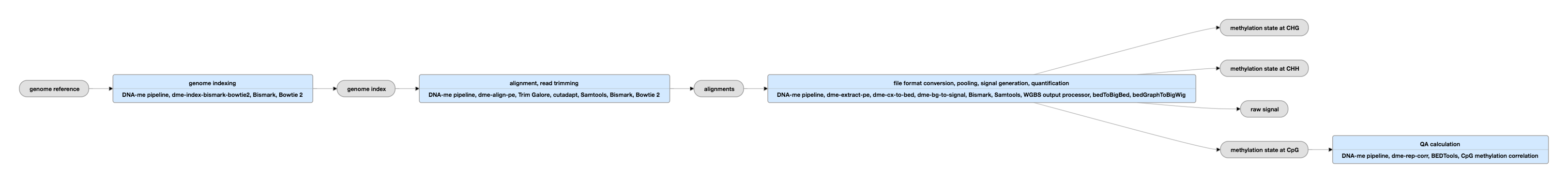
Pipeline Schematic for paired-ended data
View the current instance for paired-ended data
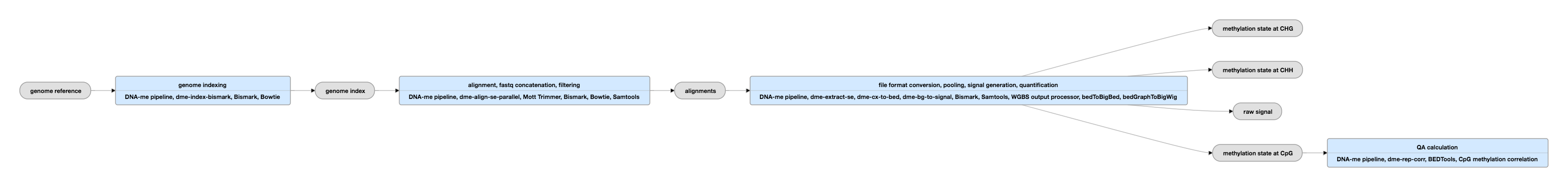
Pipeline Schematic for single-ended data
View the current instance for single-ended data
Inputs:
| File format |
Information contained in file |
File description |
Notes |
| fastq |
reads |
G-zipped DNA-sequencing reads | Reads must meet the criteria outlined under the Uniform Processing Pipeline Restrictions. |
| tar | genome index | A Bismark-transformed 2, Bowtie-indexed genome |
Outputs:
| File format |
Information contained in file |
File description |
Notes |
| bam |
alignments |
Produced by mapping reads to the genome |
Alignment software is used to produce raw bam files |
| bigWig | signal | The raw signal file of all reads | |
| bedMethyl and bigBed | methylation state at CpG | Percent methylation at CpG sites (see Description of bedMethyl file below this table). | CpG is a sequence in which a cytosine is followed by a guanine. |
| bedMethyl and bigBed | methylation state at CHG | Percent methylation at CHG sites (see Description of bedMethyl file below this table). | CHG is a sequence in which a cytosine and a guanine are separated by an adenosine, a cytosine, or a thymine. |
|
bedMethyl and bigBed |
methylation state at CHH |
Percent methylation at CHH sites (see Description of bedMethyl file below this table). |
CHH is a sequence in which a cytosine is followed by two nucleotides that may be any of adenosine, cytosine, or thymine. |
|
Quality control metrics are collected from the SamTools and Bismark software suites, and Pearson correlation is calculated from the two replicates' methylation states at CpG. |
|||
Description of bedMethyl file
The bedMethyl file is a bed9+2 file containing the number of reads and the percent methylation.
Each column represents the following:
- Reference chromosome or scaffold
- Start position in chromosome
- End position in chromosome
- Name of item
- Score from 0-1000. Capped number of reads
- Strandedness, plus (+), minus (-), or unknown (.)
- Start of where display should be thick (start codon)
- End of where display should be thick (stop codon)
- Color value (RGB)
- Coverage, or number of reads
- Percentage of reads that show methylation at this position in the genome
References
Genomic References
View the mapping assemblies (includes lambda genome for generation of comparative statistics)
View the genome annotation used in this pipeline
View the Bismark/Bowtie indices and reference files used in this pipeline
Links and Publications
Find data generated by the WGBS paired-ended pipeline
Find data generated by the WGBS single-ended pipeline
Explore WGBS-related publications on the portal here
1. Tsuji, Junko, and Zhiping Weng. "Evaluation of preprocessing, mapping and postprocessing algorithms for analyzing whole genome bisulfite sequencing data." Briefings in bioinformatics 17.6 (2015): 938-952.
2. Krueger, Felix, and Simon R. Andrews. "Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications." Bioinformatics 27.11 (2011): 1571-1572.
Uniform Processing Pipeline Restrictions
- Each replicate should have 30X coverage.
- The read length should be a minimum of 100 base pairs.
- Sequencing may be paired- or single-ended, as long as sequencing type is specified and paired sequences are indicated
- The sequencing platform must be indicated.
- Barcodes, if present in fastq, must be indicated.
- The pipeline maps against the lambda genome as a method of control.
- Alignment files are mapped to either the GRCh38 or mm10 sequences.
Current Standards
Experimental guidelines for WGBS experiments can be found here.
-
Experiments should have two or more biological replicates; they may have two technical replicates per biological replicate. Assays performed using EN-TEx samples may be exempted due to limited availability of experimental material.
- The C to T conversion rate should be ≥98%
- The CpG quantification should have a Pearson correlation of ≥0.8 for sites with ≥10X coverage.
- Sequencing may be paired- or single-ended, as long as sequencing type is specified and paired sequences are indicated.
- The experiment must pass routine metadata audits in order to be released.