ENCODE Encyclopedia Version 2:
Genomic and Transcriptomic Annotations
Introduction
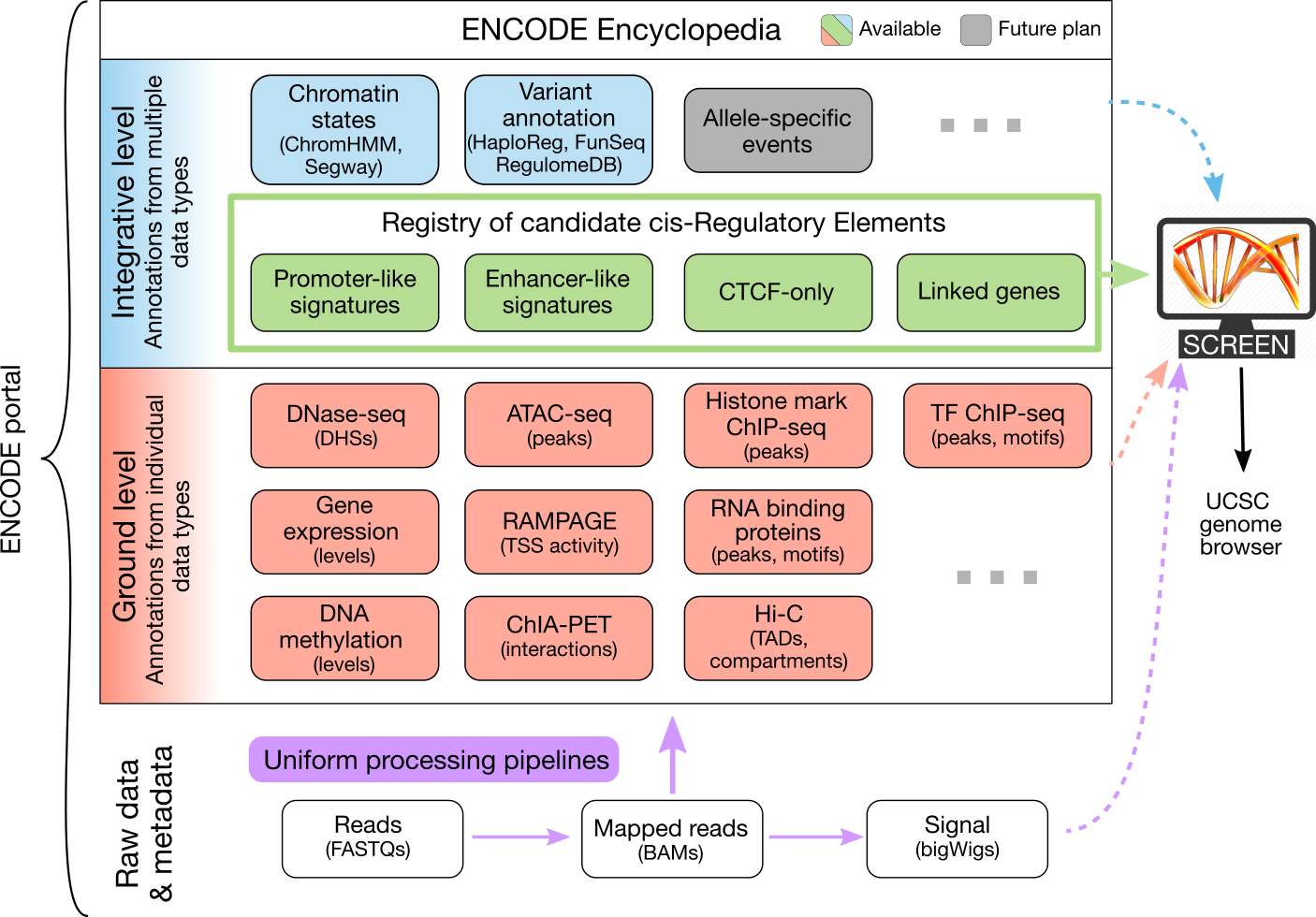
The ENCODE Consortium not only produces high-quality data, but also analyzes the data in an integrative fashion. The ENCODE Encyclopedia organizes the most salient analysis products into annotations and provides tools to search and visualize them. The Encyclopedia has two levels of annotations:
- Integrative-level annotations integrate multiple types of experimental data and ground level annotations.
- Ground-level annotations are derived directly from the experimental data, typically produced by uniform processing pipelines.
Integrative Level Annotations
The Registry of Candidate cis-Regulatory ElementsThe core of the integrative level of the ENCODE Encyclopedia is the Registry of candidate cis-Regulatory Elements (cCREs), which integrates all high-quality DNase-seq and H3K4me3, H3K27ac, and CTCF ChIP-seq data produced by the ENCODE and Roadmap Epigenomics Consortia. The cCREs in the Registry are the subset of representative DNase hypersensitivity sites (rDHSs) that are supported by these two histone modifications and CTCF-binding data. Currently the Registry (version 2) comprises 926,535 human cREs and 339,815 mouse cCREs. |
 |
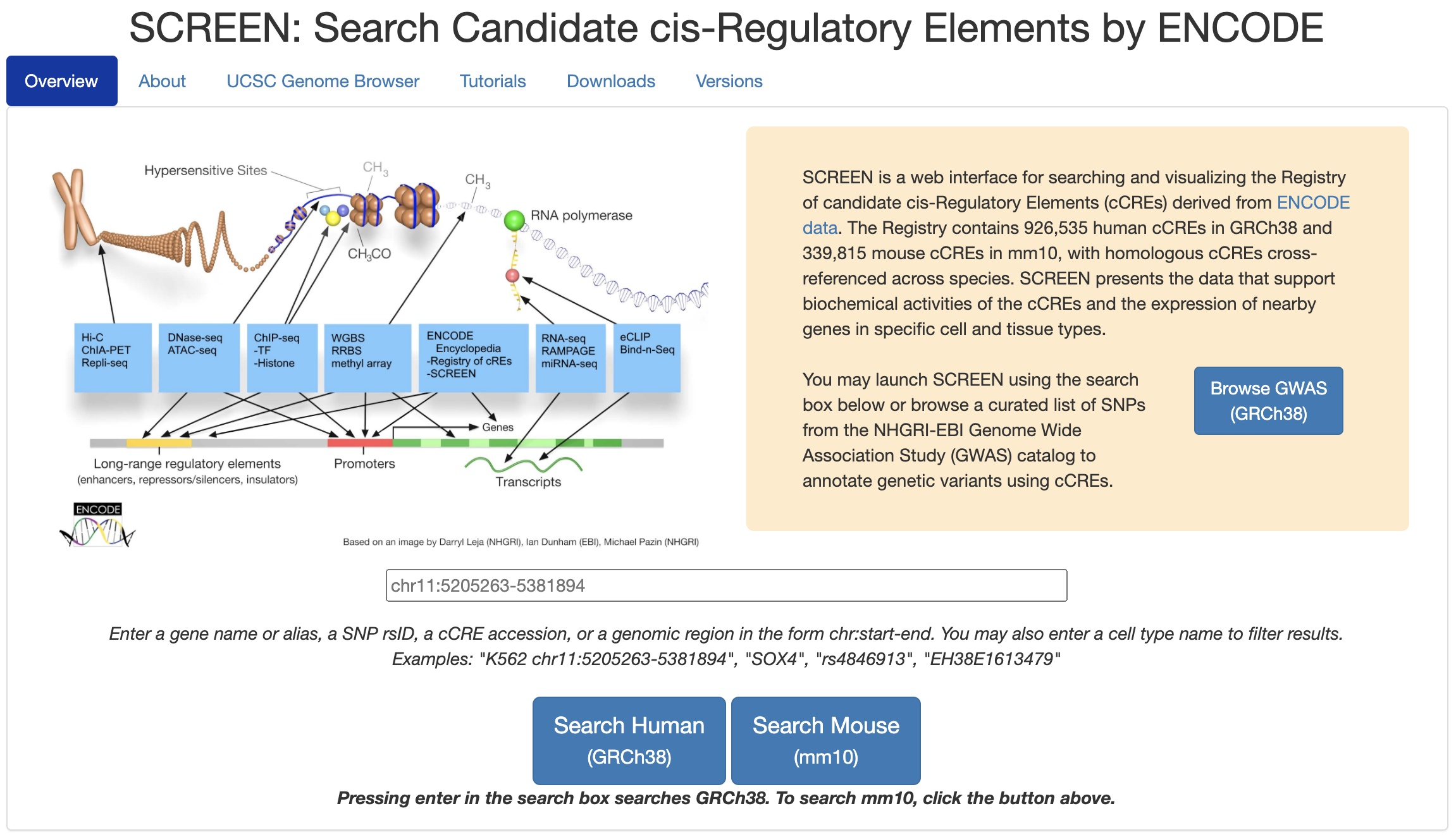
SCREENSCREEN is a web-based search and visualization engine specifically designed for the Registry of cCREs. SCREEN allows users to explore cCREs and investigate how they connect with other annotations in the Encyclopedia in a cell-type-specific manner, as well as the underlying raw ENCODE data whenever available. SCREEN also presents the results of using cCREs to interpret the variants uncovered by Genome-wide Association Studies (GWAS). [ SCREEN ] |
 |
Chromatin statesSemi-automated genomic annotation methods such as ChromHMM and Segway take as input a panel of epigenomic data (including histone mark ChIP-seq and DNase-seq) in a particular cell type and use machine learning methods to simultaneously partition the genome into segments and assign chromatin states to these segments; the states are assigned such that two segments with the same state exhibit similar epigenomic patterns. The procedure is "semi-automated" because states are then manually compared with known biological information in order to designate each state as an enhancer-like, promoter-like, gene body, etc. The chromatin states of 164 human cell types have been annotated using this strategy by integrating 1,615 genomics datasets (Libbrecht et al., (2019) Genome Biology). The chromatin states for mouse epigenomes of 12 tissue types at 8 different developmental timepoints, constituting 66 epigenomes of 8 histone marks each, have been annotated using a model integrating 1,056 genomic datasets and their respective controls. [ Chromatin states ] |
|
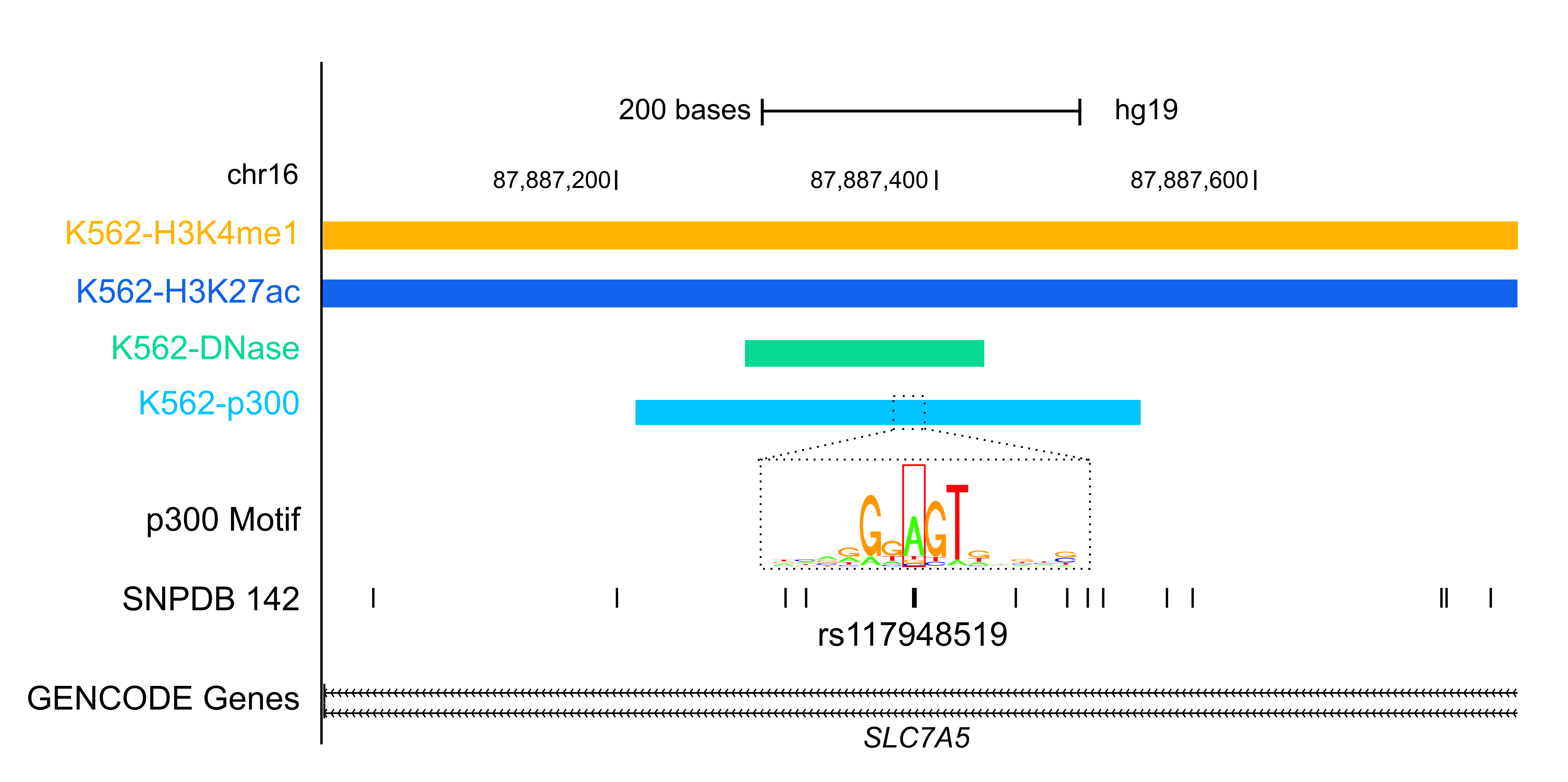
Variant AnnotationOver the past decade, Genome Wide Association Studies (GWAS) have provided insights into how genetic variations contribute to human diseases. However, over 80% of the variants reported by GWAS are in noncoding regions of the genome and the mechanism of how they contribute to disease onset is unknown. By integrating data from the ENCODE project and other public sources, RegulomeDB and HaploReg are two resources developed by ENCODE labs to aid the research community in annotating GWAS variants. FunSeq is another ENCODE resource for annotating both germline and somatic variants, particularly in the noncoding regions of cancer genomes. |
 |
Ground Level Annotations
Open chromatin (DNase-seq, ATAC-seq)DNase I hypersensitive sites (DHSs) computed from DNase-seq experiments, and ATAC-seq peaks (enriched genomic regions). |
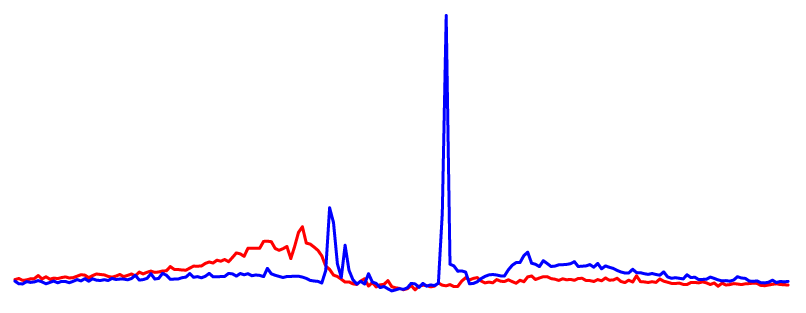 CTCF DHS Profile |
Histone mark enrichment (ChIP-seq)Peaks (enriched genomic regions) of a variety of histone marks computed from ChIP-seq experiments. |
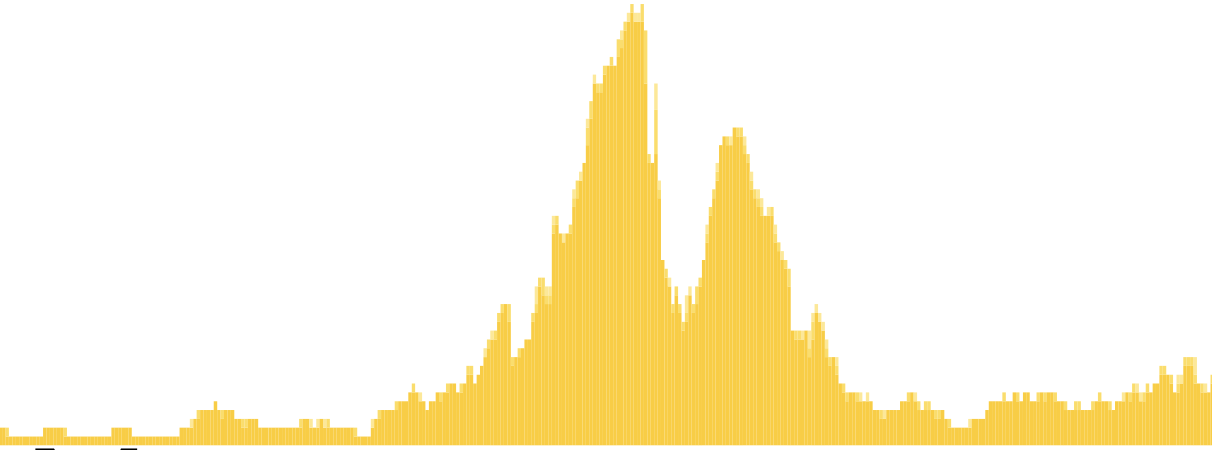 H3K27ac from mouse e11.5 hindbrain |
Transcription factor binding (TF ChIP-seq)Peaks (enriched genomic regions) of TFs computed from ChIP-seq experiments. [ TF peaks | Factorbook ] |
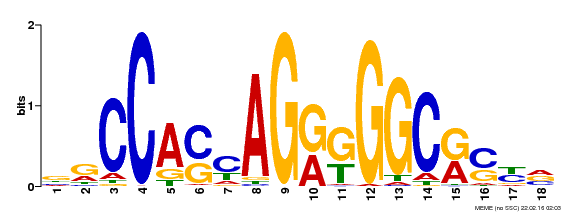 CTCF Motif from Factorbook
|
Gene expression (RNA-seq)Expression levels of genes and transcripts annotated by GENCODE, which can be visualized on SCREEN. |
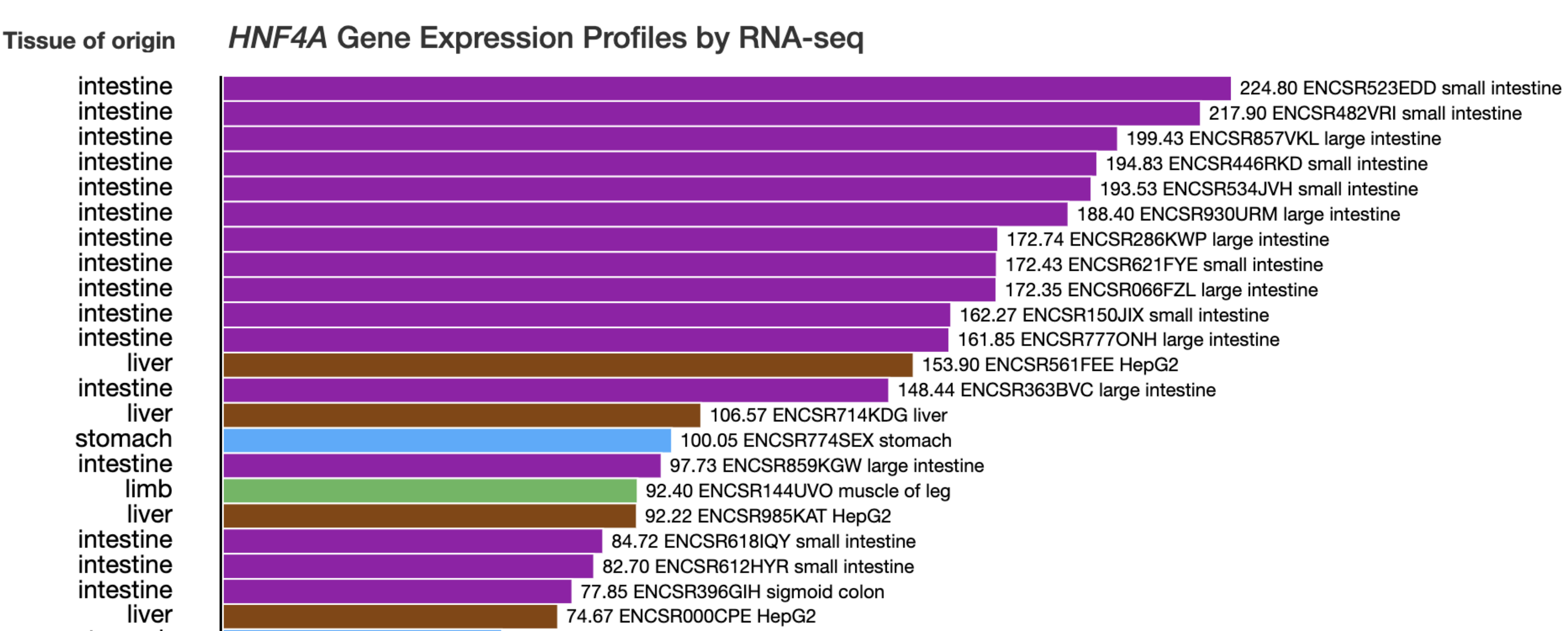 HNF4A Gene Expression |
Transcription start site (TSS) activity profiling (RAMPAGE)Identification of transcription start sites (TSSs) and quantification of transcript expression, which can be visualized on SCREEN. [ RAMPAGE peaks | SCREEN ] |
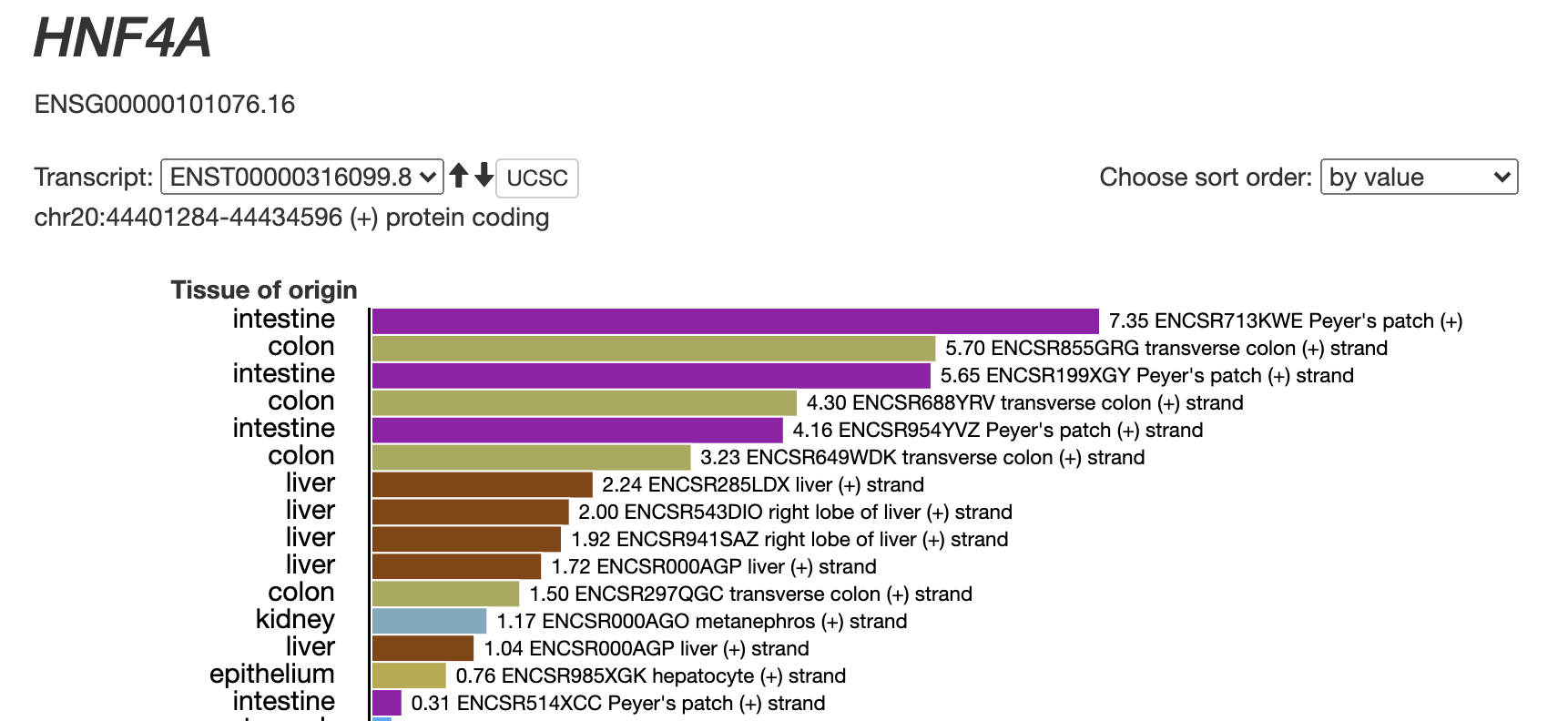 HNF4A Transcript Expression |
RNA binding protein occupancy (eCLIP-seq)Peaks (enriched genomic regions) computed from eCLIP-seq data in human cell lines K562 and HepG2 for RNA Binding Proteins (RBPs). |
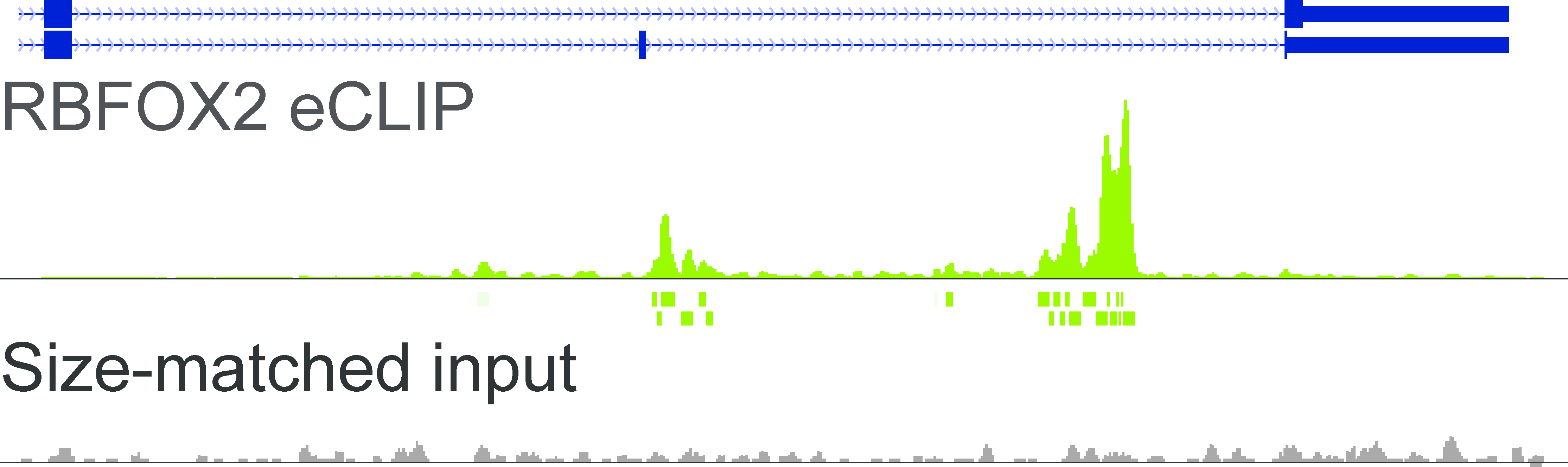 RBFOX2 read density |
DNA methylation (RRBS, WGBS)Genome-wide methylation state of CpG, CHH, and CHG dinucleotides. |
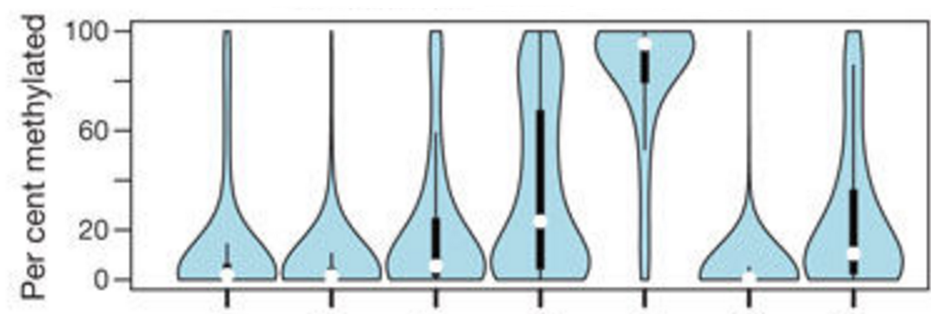 RRBS analysis in GM12878 |
Three dimensional chromatin interactions (ChIA-PET)3D interactions between genomic loci such as promoters and distal enhancers computed from ChIA-PET experiments. [ Interactions ] |
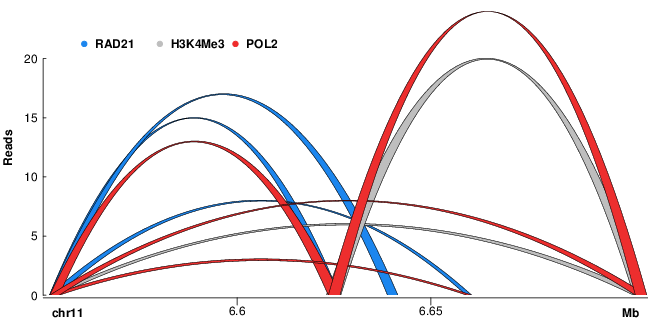 ChIA-PET interactions |
Topologically associating domains (TADs) (Hi-C)TADs and A and B compartments computed from Hi-C experiments. [ TADS | Compartments ]
|
 K562 Interaction Matrix |
Encyclopedia Versions
Ground level annotations include all released experiments to-date.
Integrative level annotations are included in V3 & V4, as indicated in their metadata.
Acknowledgements
If using annotations from the ENCODE Encyclopedia please cite the publication listed under each annotation along with:
Expanded encyclopaedias of DNA elements in the human and mouse genomes
The ENCODE Project Consortium, Moore JE*, Purcaro MJ,* Pratt HE*, Epstein CB*, Shoresh N*, Adrian J*, Kawli T*, Davis CA*, Dobin A*, Kaul R*, Halow J*, Nostrand EL*, Freese P*, Gorkin DU*, Shen Y*, He Y*, Mackiewicz M*, Pauli-Behn F*, Williams BA, Mortazavi A, Keller CA, Zhang X, Elhajjajy S, Huey J, Dickel DE, Snetkova V, Wei X, Wang X, Rivera-Mulia JC, Rozowsky J, Zhang J, Chhetri SB, Zhang J, Victorsen A, White KP, Visel A, Yeo GW, Burge CB, Lécuyer E, Gilbert DM, Dekker J, Rinn J, Mendenhall EM, Ecker JR, Kellis M, Klein RJ, Noble WS, Kundaje A, Guigó R, Farnham PJ, Cherry JM†, Myers RM†, Ren B†, Graveley BR†, Gerstein MB†, Pennacchio LA†, Snyder MP†, Bernstein BE†, Wold B†, Hardison RC†, Gingeras TR†, Stamatoyannopoulos JA†, Weng Z†
Nature, 30 July 2020
* Authors contributed equally
† Corresponding authors
Data from the Common fund supported Roadmap Epigenomics Mapping Consortium (REMC) were included for building the ENCODE Encyclopedia. Please see the 2015 paper on their analysis of reference human genomes for more information.