ChIA-PET Data Standards and Prototype Processing Pipeline
Assay overview
ChIA-PET is a method for capturing genome-wide chromatin interactions that involve a protein of interest. First, protein-DNA interactions are stabilized by dual-cross-linking in cells. Then, nuclei are released by cell lysis and are sonicated to generate chromatin complexes containing DNA fragments. Immunoprecipitation (IP) is performed using a specific antibody to enrich for chromatin complexes involving a protein of interest. Chromatin complexes immobilized on antibody beads are then subjected to DNA end repair and A-tailing. Next, pairs of DNA fragments are joined by proximity ligation with a “bridge linker” – a short double-stranded DNA sequence containing an internal biotinylated nucleotide and T overhangs on each end. The ligated DNA fragments are released by reverse cross-linking, and Tn5 transposase is used to simultaneously fragment the DNA and add sequencing adaptors. Streptavidin beads are then used to enrich for DNA fragments containing ligation junctions (i.e., containing the biotinylated bridge linker). These fragments are subjected to PCR amplification with minimal cycles, size selection of the DNA fragments, and high-throughput paired-end sequencing. To analyze the data, reads pairs are first partitioned into three categories: (i) read pairs with no linker sequence, (ii) read pairs with a linker sequence and one usable genomic tag, or (iii) read pairs with a linker sequence and paired end tags (PETs). Each category is then aligned to a reference genome and only uniquely mapped and non-redundant tags are retained for further analysis. The final PETs are used to generate a 2D contact-map file for visualization and also to identify clusters of overlapping intrachromosomal loops. The final tags (from all three categories of read pairs) are used to identify genomic binding sites of the protein of interest via peak calling. Haplotype-resolved chromatin interactions can be deduced if phased SNP information is available for the appropriate reference genome. (Li. et al. Long-read ChIA-PET for base-pair resolution mapping of haplotype-specific chromatin interactions. Nat Protoc. 2017 May; 12(5): 899–915.)
Pipeline Overview
The ChIA-PET pipeline (ChIA-PIPE) was developed by the Jackson lab. The full ChIA-PET pipeline code is available on Github.
ChIA-PIPE is a fully automated pipeline for ChIA-PET data processing, quality assessment, visualization, and analysis. ChIA-PIPE performs linker filtering, read mapping, peak calling, and loop calling and automates quality control assessment for each dataset. To enable visualization, ChIA-PIPE generates input files for two-dimensional contact map viewing with Juicebox and HiGlass and provides a new dockerized visualization tool for high-resolution, browser-based exploration of peaks and loops. To enable structural interpretation, ChIA-PIPE calls chromatin contact domains, resolves allele-specific peaks and loops, and annotates enhancer-promoter loops. ChIA-PIPE also supports the analysis of other related chromatin-mapping data types.
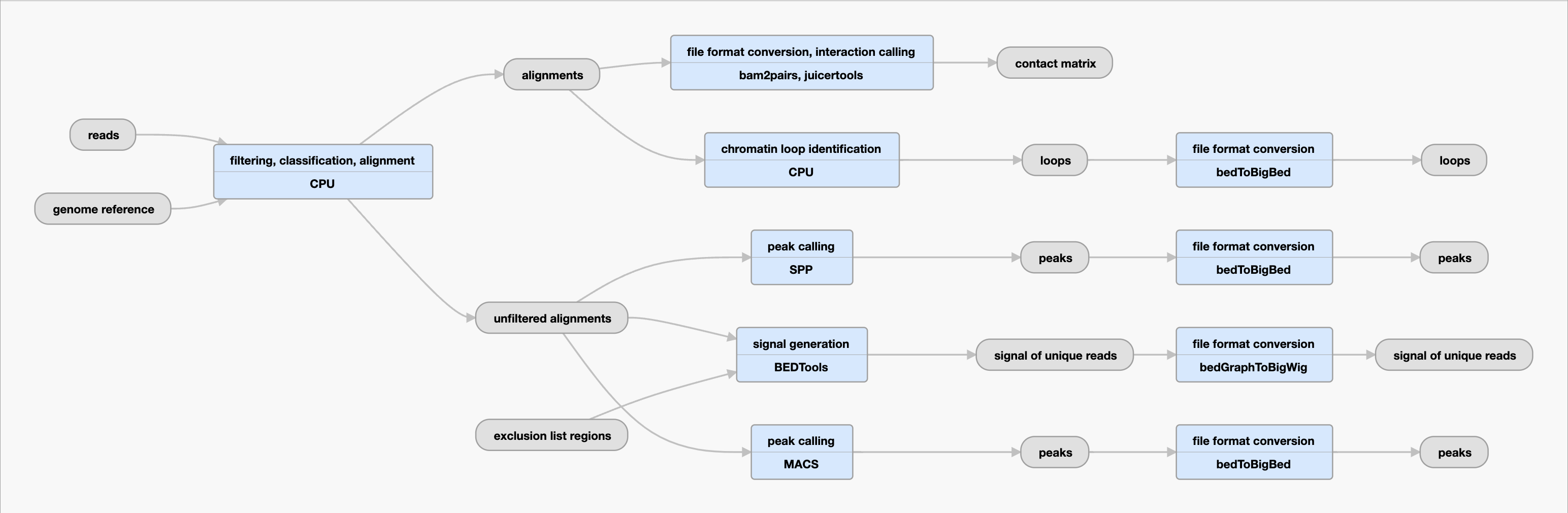
Inputs:
| File format |
Information contained in file |
File description |
Notes |
| .fastq | reads | G-zipped, paired-ended reads. | If multiple fastq files are generated, then fastq files are merged. |
| .fasta | text-based format | reference genome sequences | GRCh38 for human, mm10 for mouse |
Outputs:
| File format |
Information contained in file |
File description |
Notes |
| .bam | alignments | Produced by trimming sequencing adaptors, identifying the ChIA-PET bridge linker sequence from input reads, collecting singlelinker 2 tags reads, mapping these filtered reads to the reference genome, quality filtering, deduplicating, and sorting. | These reads are ChIA-PET single linker 2 tags, meaning two qualified genomic sequences connected with a linker, where both genomic sequences is larger than or equal to 18bp. The singlelinker 2 tags are used for chromatin interactions and long range chromatin interactions. |
| .bedgraph | signal of unique reads | Produced by bedtools genomecov using unfiltered alignments. | It filters exclusion list regions. |
|
.bed .bigBed (narrowPeak) |
peaks | Produced by MACS2 (narrowpeak) using unfiltered alignments. BigBed is produced by KentUtils (bedToBigBed) using a peak file. | The pipeline uses MACS2, if there is no input control ChIP-seq. When KentUtils produces bigBed file, MACS2 uses narrowpeak (bed6+4). MACS2 bigBed is truncated score to 1000. |
|
.bed .bigBed (broadPeak) |
peaks | Produced by SPP (broadpeak) using unfiltered alignments. BigBed is produced by KentUtils (bedToBigBed) using a peak file. | The pipeline uses SPP, if there is an input control ChIP-seq. SPP called peaks contain less false-positive peaks, compared with MACS2 called peaks.When KentUtils produces bigBed file, SPP peak uses broadpeak (bed6+3). |
| .bedpe | loops | Produced by CPU cluster using uniquely mapped and deduplicated singlelinker 2 tags reads. The pipeline collects intra-chromosomal interactions (filtering out inter-chromosomal interactions), and also filter out noisy random interactions based on PET count score. | Uniquely mapped and deduplicated singlelinker 2 tags reads are piled up. When both ChIA-PET anchors are overlapped, it is merged, and counting merged number as PET-count score. The pipeline filters out noisy random interaction where PET count score is less than or equal to 2. |
| .bigInteract | loops |
bigInteract is produced by KentUtils (bedToBigBed) using a bedpe file. |
When KentUtils produces bigInteract file, it uses bed file format (bed5+13) with interact option. bigInteract is truncated PET count score to 1000. |
| .bigWig | signal of unique reads | bigWig is produced by KentUntils (bedGraphToBigWig) using a bedgraph file. | |
| .hic | contact matrix | hic file is produced by Juicertool (pre) using pairs file, which is generated from bam2pairs. It uses uniquely mapped and deduplicated singlelinker 2 tags reads. | It contains both intra-chromosomal interactions and inter-chromosomal interactions. It does not filter out based on PET count score, meaning that contact map (hic) shows all interaction data. |
Current Standards
Experimental and Computational guidelines for ChIA-PET experiments can be found here.
The experiments must pass routine metadata audits in order to be released. Here are the quality metrics and their standards:
- In the alignment quality metric:
- total read pairs recommended ≥ 150,000,000, high quality ≥ 180,000,000
- fraction of read pairs with bridge linker recommended ≥ 0.5
- number of non-redundant PET recommended ≥ 10,000,000
- In the chromatin interactions quality metric:
- ratio of intra/inter-chr PET recommended ≥ 1
- In the peak enrichment quality metric:
- number of protein factor binding peaks recommended ≥ 10,000