eCLIP Data Standards
Assay Overview
eCLIP is an enhanced version of the crosslinking and immunoprecipitation (CLIP) assay, and is used to identify the binding sites of RNA binding proteins (RBPs). Because the CLIP and iCLIP methods often result in high duplication rates and low library complexity, eCLIP seeks to improve the efficiency and quality of library production (van Nostrand, et. al, Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP), Nature Methods (2016),13: 508–514
Updated June 2017
Pipeline Schematic
View current instance of this pipeline
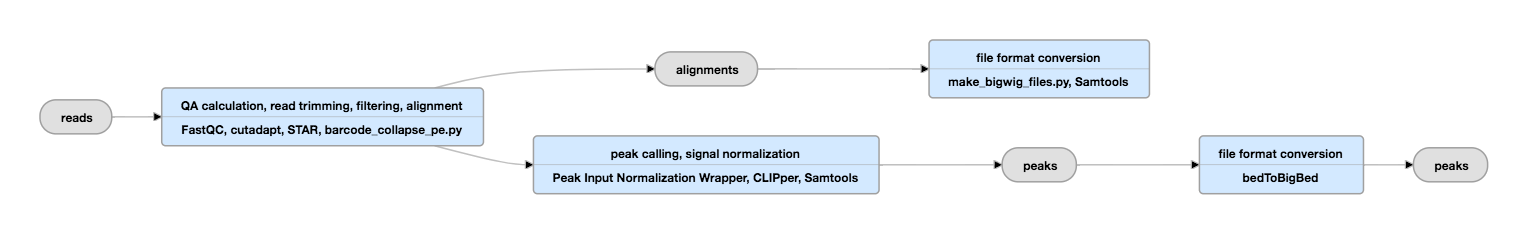
Find data generated by this pipeline here
Updated June 2017
ENCODE 3 Standards
Experimental guidelines for eCLIP experiments can be found here.
- Experiments should have two or more biological replicates, isogenic or anisogenic.
- Antibodies must be characterized according to standards set by the ENCODE Consortium. Please see the linked documents for transcription factor standards (May 2016), histone modification and chromatin-associated protein standards (October 2016), and RNA binding protein standards (November 2016).
- Each eCLIP-seq experiment should have a corresponding size-matched input control experiment, with matching run type and read length.
- eCLIP experiments should have 1 million unique fragments or have saturated peak detection in each biological replicate.
- Replicate concordance is measured by calculating IDR values (Irreproducible Discovery Rate). The experiment passes if both rescue and self consistency ratios are less than 2.
- Narrow binding RBPs should have a FRiP score over enriched peaks of at least 0.005.
- RBPs with non-typical binding patterns (including binding to rRNA, snRNAs, or other repetitive elements or multicopy sequences, broad binding to entire transcripts or transcript regions, or binding to specific RNA families) may pass via exemption.
- The experiments must pass routine metadata audits in order to be released.
Uniform Processing Pipeline Restrictions
- The read length must be 50 base pairs.
- Sequencing must be paired-ended.
- The sequencing platform used should be indicated.
- Replicates should match in terms of read length and run type.
- Pipeline files are mapped to GRCh38, hg19, or mm10 sequences.
- Gene and transcript quantification files are annotated to GENCODE V24 (GRCh38), V19 (hg19), or M4 (mm10).