Long Read RNA-seq Data Standards and Processing Pipeline
Assay Overview
Alternative splicing is widely acknowledged to be a crucial regulator of gene expression and is a key contributor to both normal developmental processes and disease states. Long read RNA-seq can sequence individual transcript molecules in full, making it possible to see exactly which splice junctions were present in context. Long read RNA-seq can be used to characterize isoforms on a transcriptome-wide scale, and also to perform quantification on the gene and isoform level.
Pipeline overview
The ENCODE long read RNA-seq pipeline can be used for PacBio or Oxford Nanopore libraries generated from full length cDNA/RNA transcripts with a poly-(A) tail. For effective quantification, see the read depth requirements outlined in the Current Standards section. The long red RNA-seq pipeline was developed by Ali Mortazavi's group at the University of California, Irvine.
Pipeline Schematic
View the current instance of this pipeline on the ENCODE Portal.
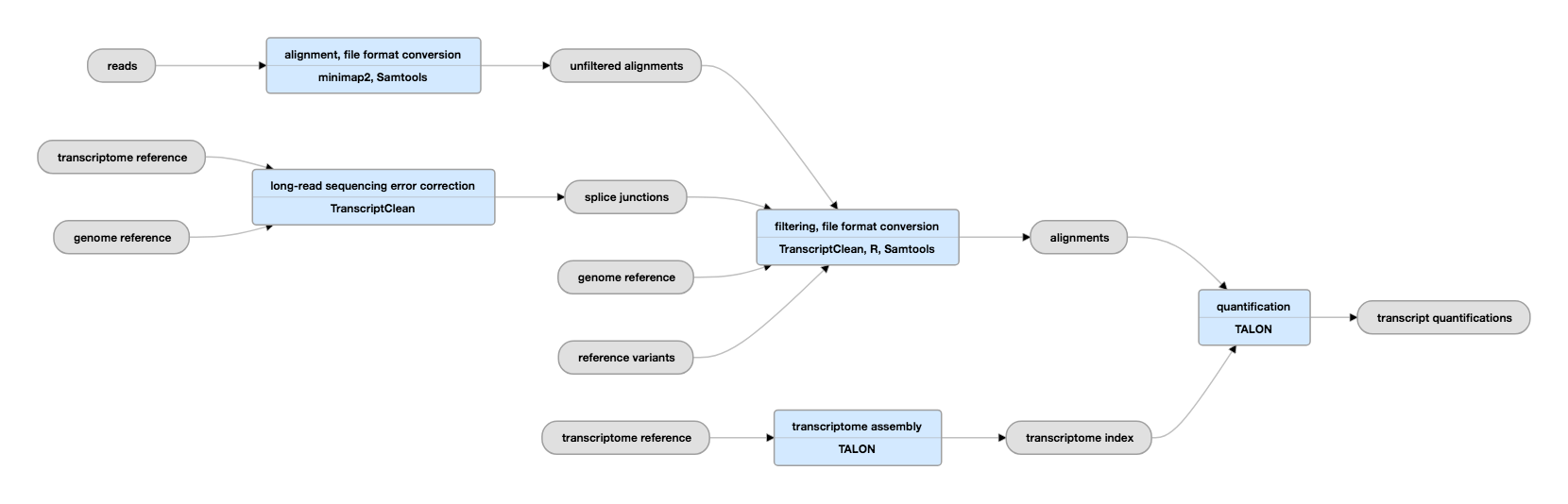
Inputs:
| File format |
Information contained in file |
File description |
Notes |
| fastq |
reads |
Full-length long read RNA-seq reads. For PacBio: Circular-consensus processed reads with adapters and poly-(A) tail removed. |
Reads must meet the criteria outlined in the Uniform Processing Pipeline Restrictions. |
| database, tsv, fasta, gtf |
reference files |
Default reference genome, transciptome annotation file from GENCODE, and variant file from dbSNP. | See the References section below for details. |
Outputs:
| File format |
Information contained in file |
File description |
Notes |
| bam | unfiltered alignments | Produced by mapping reads to the genome. |
This file contains the original Minimap2 alignments. No modifications have been made to the read sequences at this point. |
| bam | alignments | Produced by performing reference-based error correction of the unfiltered alignments (TranscriptClean). | Reads that contain one or more un-annotated noncanonical splice junctions are removed, as are any non-primary read alignments. |
| tsv | transcript quantifications | Non-normalized transcript counts. | |
| The pipeline also produces quality metrics, including Spearman correlation and read depth. | |||
References
Obtaining Full-Length Reads
Before aligning long read RNA-seq reads to the genome, it is important to apply platform-specific steps to make sure that the reads are full-length and to trim off adaptors. See protocols for human or mouse samples to see further details for performing circular consensus correction on raw PacBio subreads, and for trimming adaptors and poly-(A) tails from the PacBio circular consensus output.
Genomic References
View the mapping assembly and genome annotation reference files used in this pipeline.
Additional reference files
View the reference variants, splice junctions, and TALON.db files used in this pipeline.
View the SIRV spike-in reference files used in the long read RNA-seq datasets.
Links and Publications
View the GitHub repo for the ENCODE long-read RNA-seq pipeline.
Uniform Processing Pipeline Restrictions
- Read length should be a minimum of 300 base pairs.
- PacBio and Oxford Nanopore long read platforms are supported by this pipeline.
- Alignment files are mapped to either the GRCh38 or mm10 sequences.
- Gene and transcript quantification files are annotated to either GENCODE V29 or M21.
Current Standards
-
Experiments should have two or more replicates. Assays performed using EN-TEx samples may be exempted due to limited availability of experimental material.
- Experiment must pass routine metadata audits in order to be released.
| Resulting data status | Sequencing depth | Mapping rate |
Replicate concordance (Spearman correlation) |
Number of GENCODE genes detected |
|---|---|---|---|---|
| Good | ≥ 600,000 FLNC* reads/replicate | ≥ 90% mapped reads | ≥ 0.8 | ≥ 8,000 genes detected/replicate |
| Acceptable | 400,000-600,000 FLNC reads/replicate | 60-90% mapped reads | 0.6-0.8 |
4,000-8,000 genes detected/replicate** |
| Poor | < 400,000 FLNC reads/replicate | < 60% mapped reads | < 0.6 |
< 4,000 genes detected/replicate** |
|
* full-length nonchimeric reads ** Exemptions may be made for samples where supporting short read RNA-seq data is available to validate the low gene count, given that short read samples are from the same cell type/cell line and pass all ENCODE standards |
||||