microRNA-seq Data Standards and Processing Pipeline (E4)
Assay Overview
MicroRNA-seq allows researchers to characterize and quantify the expression and prevalence of the small non-coding RNA moleccules known as microRNA. These molecules may play an important role in diseases, and significant effort is underway to understand their effects across a variety of tissue types and cells. For effective processing, the average insert size must be no more than 30 bases.
Updated May 2017
Pipeline Overview
The ENCODE miRNA-seq pipeline can be used for libraries generated from miRNAs, size-selected from total RNA to be 30 bp or smaller. The microRNA-seq pipeline was developed by Ali Mortazavi's group at UC Irvine.
Pipeline Schematic
View the current instance of this pipeline.
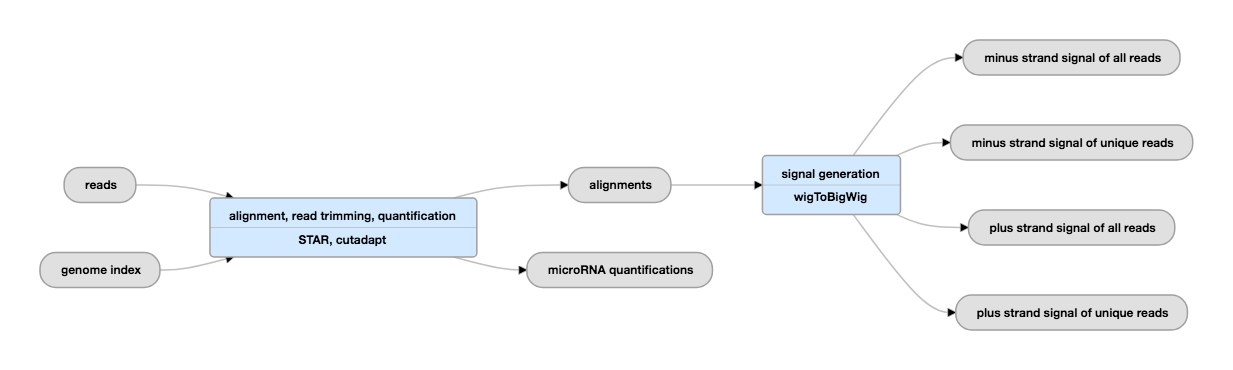
Inputs:
| File format |
Information contained in file |
File description |
Notes |
| fastq |
reads |
microRNA-seq reads from single-end libraries. | Reads must meet the criteria outlined in the Uniform Processing Pipeline Restrictions. |
| gtf, tar | reference files | Genome annotations, adapter sequences, genome indices | See the References section below for details. |
Outputs:
| File format |
Information contained in file |
File description |
Notes |
| bam |
alignments |
Produced by mapping reads to the genome. | Reads are trimmed using a proprietary version of cutAdapt, linked below under References. |
| bigWig | signal | Normalized RNA-seq signal | Signals are generated for both the plus and minus strands and for unique reads and unique+multimapping reads. |
| tsv | gene (microRNA) quantifications | Non-normalized counts. | |
| Quality control metrics are also generated, including gene quantification level and read depth. | |||
The mapping of the reads is done using the STAR aligner. STAR is also used to obtain counts of miRNAs (number of reads mapped to each miRNA gene in the annotations file). The miRNA counts can be normalized, for example by library size, to obtain counts-per-million for downstream analysis.
References
Adapter Trimming
An important step before aligning miRNA-seq reads is the trimming of adapters. The raw sequencing reads for samples generated for this pipeline contain 3’ and 5’ adapters. The links below contain details of the pipeline including information about the adapter sequences used and an example script to trim these adapters:
-
View the adapter sequences and an example Cutadapt script (version 1.7.1 used to generate data by this pipeline) to trim the adapter sequences.
- View the index generation step (using comprehensive GENCODE annotations) and the alignment step of trimmed reads Using STAR (using miRNA subset of GENCODE annotations).
Genomic References
View the mapping assembly and genome annotation reference files used in this pipeline.
STAR Indices
This pipeline requires both assembly information for the species of interest and a gene reference. STAR creates an index for use in the mapping step. Please note that the comprehensive GENCODE references are used for the index generation step, while the miRNA subsets of the corresponding annotations are used at the alignment step to obtain miRNA counts by STAR.
The miRNA subset of GENCODE V29 for human (used at the mapping step by STAR)
The miRNA subset of GENCODE M21 for mouse (used at the mapping step by STAR)
Links and Publications
Find data generated by the pipeline here.
Uniform Processing Pipeline Restrictions
- The mapped read length should be a minimum of 16 base pairs.
- If paired-ended data is used as input, the pipeline will only process read 1.
- All Illumina platforms are supported for use in the uniform pipeline; colorspace (SOLiD) are not supported.
- The microRNAs protocol involves a gel size selection of the fragments that correspond to insert length < 30.
- Alignment files are mapped to either the GRCh38 or mm10 sequences.
- Gene and transcript quantification files are annotated to either GENCODE V29 or M21.
Current Standards
- Experiments should have two or more replicates. Assays performed using EN-TEx samples may be exempted due to limited availability of experimental material.
- The experiment must pass routine metadata audits in order to be released.
| Resulting data status | Sequencing depth | Replicate concordance (Spearman correlation) | Number of miRNAs expressed |
|---|---|---|---|
| Good |
≥ 5 million reads mapped to the miRNA subset of genome (unique + multimap < 10) |
≥ 0.85 | ≥ 300 miRNAs at minimum 2cpms per replicate |
| Acceptable | 3-5 million reads mapped to the miRNA subset of the genome (unique + multimap < 10) in either replicate | 0.8-0.84 |
200-300 miRNAs at minimum 2cpms in either replicate* |
| Poor | < 3 million reads mapped to the miRNA subset of the genome (unique + multimap < 10) in either replicate | < 0.8 |
< 200 miRNAs at minimum 2cpms in either replicate* |
| * Exemptions may be made for samples with higher concordance (Spearman correlation > 0.85) or deeper sequencing depth (> 5M) in the case of EN-TEx samples with one replicate, or in the case of known low gene counts from short RNA-seq data obtained from the same cell line/tissue which has passed ENCODE standards. | |||