microRNA Counts Data Standards and Processing Pipeline
Assay Overview
The ENCODE Nanostring pipeline for microRNAs (miRNA) can be used for nCounter miRNA Reporter Code Count (RCC) datasets to generate normalized miRNA counts in tabular, bed, and bigBed formats.
Updated May 2017
Pipeline Overview
The microRNA counts pipeline was developed by Ali Mortazavi's group at UC Irvine.
Pipeline Schematic
View the current instance of this pipeline
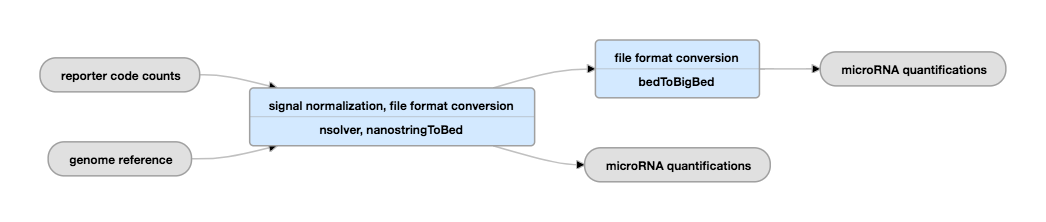
File inputs and outputs:
| File format |
Information contained in file |
File description |
Notes |
| rcc |
raw counts |
Reporter Code Counts, containing the counts for each gene. | This is an input file for the pipeline. |
| gff/gff3 | gene annotation | Default gene annotation file is from mirRBase. | This is an input file for the pipeline. |
| bed, bigBed | miRNA quantifications | Normalized counts formatted for track display. | This is an output file from the pipeline. |
| Quality control metrics are also generated, including gene quantification level and read depth. | |||
Normalization of miRNA counts:
Raw miRNA counts in RCC format are processed using nSolver Analysis Software v2.5. The detailed steps are:
- RLF files for the corresponding data sets is uploaded to nSolver Analysis Software. The RLF files are proprietary, but this set of data uses the Human v2.1, Mouse v1.2, Mouse v1.3, and Mouse v1.5 platforms.
- Raw miRNA counts in RCC format are uploaded to nSolver Analysis Software.
- The 2 biological replicates of a sample are selected.
- The maximum background level (negative controls) is subtracted from the sample counts.
- The samples are normalized by the geometric means of the positive controls and the top 100 expressed miRNAs.
- The results are saved in tab-delimited text format with four columns:
- Gene Name
- Accession #
- Replicate 1 counts
- Replicate 2 counts
References
Genomic References
View the reference files used in this pipeline
View the mirBase references for mouse and human
Links and Publications
Find data generated by this pipeline here
View the custom script to convert normalized counts to bed
Uniform Processing Pipeline Restrictions
- Normalized counts in bed format are mapped to either the GRCh38 or mm10 sequences.
- Gene and transcript quantification files are annotated to miRBase V21 (mouse, human).
- All Nanostring nCounter miRNA Reporter Code Count (RCC) files for human and mouse are supported for use in the uniform pipeline.
Current Standards
- Experiments should have two or more replicates. Assays performed using EN-TEx samples may be exempted due to limited availability of experimental material
- The normalized counts for the two biological replicates of a sample should have a Pearson correlation of ≥0.90.
- The experiment must pass routine metadata audits in order to be released.