HiC Data Standards and Processing Pipeline
Assay Overview
Hi-C is an assay that uses DNA-DNA proximity ligation to identify long range chromatin contacts in an unbiased, genome-wide fashion. This gives insight about the 3D architecture of the genome inside the nucleus. In a typical Hi-C assay, cells are fixed, causing loci in close spatial proximity to be cross-linked with one another. Next, the DNA is fragmented, and sequences that are nearby in 3D are ligated to one another. The resulting chimeric sequences capture information about the DNA sequences that were co-localized in the original sample. The sequence of the ligation junction can be used to determine the adjacent positions with single base-pair resolution. Hi-C libraries are characterized by means of DNA sequencing, generating a library of chimeric sequences that were in close proximity within the nucleus. This catalog of chimeric sequences can be used to construct a contact matrix showing the frequency with which pairs of loci are in contact.
Analysis of Hi-C data reveals many features of genomic organization, including compartments (Lieberman-Aiden E. et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009 Oct 9;326(5950):289-93.), subcompartments, contact domains, and point-to-point chromatin loops (Rao, Huntley et al. A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping. Cell. 2014 Dec 18; 159(7):p1665-1680.), as well as stripes in which a single locus tends to form contact with a genomic interval (Vian L, et al. Casellas R. The Energetics and Physiological Impact of Cohesin Extrusion. Cell. 2018 May 17;173(5):1165-1178.e20.).
Hi-C data can also be used for genotyping, both to call variants (including local variants, such as SNPs, as well as large scale structural variants), and to assign them to chromosome-length haploblocks (Dudchenko O, 3D genomics across the tree of life reveals condensin II as a determinant of architecture type. Science. 2021 May 28;372(6545):984-989.).
Updated September 2022
Pipeline Overview
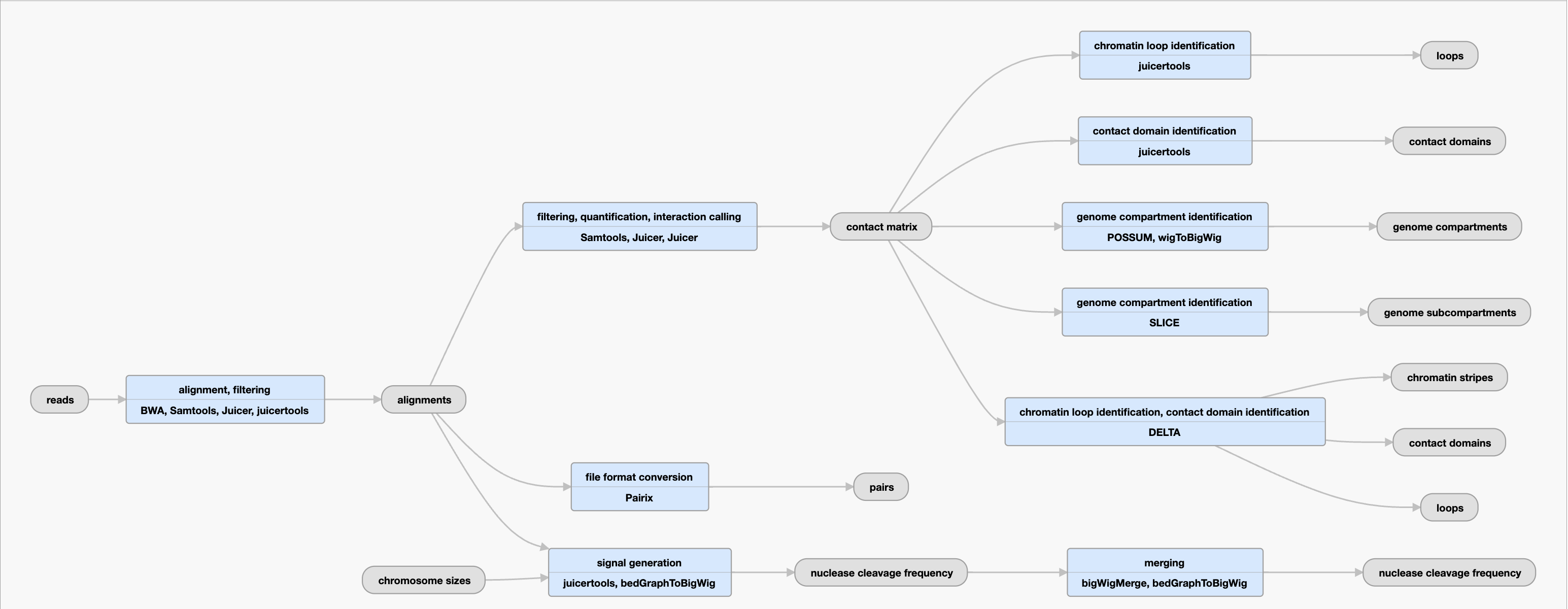
The Hi-C pipeline was developed by Aiden lab. The full Hi-C pipeline code is available on Github.
Inputs:
| File format |
Information contained in file |
File description |
Notes |
| .fastq | reads | G-zipped reads, paired-ended or single ended, stranded or unstranded. | Multiple fastqs from a single biological replicate or library are concatenated before mapping. Reads must meet the criteria outlined under the Uniform Processing Pipeline Restrictions. |
| .tsv | chromosome sizes | Reference file (GRCh38, mm19 available) used in the ENCODE Processing Pipeline | |
| .txt | restriction enzyme map | Map of restriction enzyme sites across the genome (GRCh38, mm19 available) | Applicable only for experiments using restriction enzymes. |
Outputs:
| File format |
Information contained in file |
File description |
Notes |
| .bam | alignments | Filtered and de-duplicated bam file produced by mapping reads to the genome | Reads aligned to GRCh38 (for experiments in human) or mm10 (for experiments in mouse) |
| .bedpe | contact domains | Chromatin intervals that exhibit enhanced contact frequency within themselves | May include multiple files with this output depending on if the file was derived_from mapq1 or mapq30. The preferred files to use for analysis are those with mapq30 annotations. They are marked with preferred_default=true property. |
| .bigWig | genome compartments | A-B compartments, which result from the spatial segregation of open and closed chromatin to form two genome-wide compartments. Positive values correspond to the A compartment. | May include multiple files with this output_type that differs in bin size (i.e. 5kb, 10 kb). The preferred files to use for analysis are those with mapq30 annotations. They are marked with preferred_default=true property. |
| .hic | mapping quality thresholded contact matrix | A contact matrix after filtering out all alignments with mapQ <30 | The preferred files to use for analysis are those with mapq30 annotations. They are marked with preferred_default=true property. |
| .hic | contact matrix |
By partitioning the linear genome into “loci” of fixed size (e.g., bins of 1 Mb or 1 kb), the Hi-C map can be represented as a “contact matrix” M, where the entry Mi,j is the number of contacts observed between locus Li and locus Lj.
This file contains contact matrices at resolutions as fine as 10bp.
|
To account for nonuniformities in coverage due to the number of restriction sites at a locus or the accessibility of those sites to cutting, we use a matrix-balancing algorithm to generate a normalization for each chromosome and resolution; these are also included in the file. |
| .pairs | pairs | This file contains a list of all Hi-C contacts: reads (or read pairs) reflecting at least two ligated sequences that align uniquely to the genome. Duplicates are removed. | |
| .bigWig | nuclease cleavage frequency | Frequency of nuclease cutting observed for each genomic position. Correlates with chromatin accessibility in high-quality samples. | |
| .bed | genome subcompartments | Patterns of long-range contacts | |
| .bedpe | loops | Pairs of loci that show significantly closer proximity with one another than with the loci lying between them | May include multiple files with this output depending on if the file was derived_from mapq1 or mapq30.The preferred files to use for analysis are those with mapq30 annotations. They are marked with preferred_default=true property. |
Current Standards
The experiment must pass routine metadata audits in order to be released. The chromatin interaction (.hic) is checked for the following quality metrics:
Intact Hi-C Experiments
- minimum of 2 billion total reads
- minimum of 50% unique reads in Hi-C contacts and ideally have a percentage over 60%. A low percentage of unique reads in Hi-C contacts indicates a failure in the restriction, fill-in, or ligation steps of the protocol
- minimum of 15% of unique reads in long range contacts
In situ Hi-C Experiments
- a maximum of 40% of unique total duplicates. A high percentage of unique total duplicates indicates low molecular complexity
- require a minimum 20% of unique reads in Hi-C contacts, and ideally have a percentage over 50%. A low percentage of unique reads in Hi-C contacts indicates a failure in the restriction, fill-in, or ligation steps of the protocol
- a minimum 15% of reads with ligation motif present. A low percentage of reads with ligation motif present indicates a failure in ligation
- a minimum 20% of unique reads in long range contacts, and ideally have a percentage over 35%
- Experimental guidelines for in situ HiC experiments can be found here
- Computational guidelines for in situ HiC experiments can be found here