Data Submission
Project overview | Using the portal | REST API | Data organization | Data submission | Encyclopedia
Welcome
Welcome to the ENCODE Portal! The Data Coordination Center (DCC) have prepared documentation to assist you with submitting your data to the Portal. Providing reliable metadata is essential for maintaining the high standard of quality set by the ENCODE Consortium and making the Portal a reliable and valuable resource for the scientific community as a whole.
We achieve this by representing discrete elements of an experiment or project as unique JSON objects on the Portal, each with their own identifying metadata properties. This means that every time you submit an object, it receives a unique identifier: a uuid. For some of the objects (such as Donor, Biosample, Library, Experiment, and more), an accession (following the format ENC[SR|BS|DO|GM|AB|LB|FF|PL][0-9]{3}[A-Z]{3} where [SR|BS|DO|GM|AB|LB|FF|PL] refer to the metadata type given the accession) is also assigned. Though these are automatically generated, you also have the opportunity to assign an identifier to your objects with something unique that makes sense to you. This unique identifier is specified in the aliases property. The aliases identifiers follow the format of the lab name followed by a colon and your chosen identifier (e.g., john-doe:experiment_01, assuming your lab name is john-doe). These three types of IDs (uuid, accession, and alias) can be used interchangeably to refer to an object. For a more complete introduction to our data model, accessions, and ontologies, please read this page.
NOTICE: If you are preparing to submit data to the ENCODE Portal, you should already have a user account, be associated with a lab and grant/award, and been given API access keys. If you are not sure or are missing any of these things, please contact your data wrangler at the DCC. If you don't know who your data wrangler is, please contact the help-desk at encode-help@lists.stanford.edu.
Submission setup
Setting up your tools
First, follow the instructions for installing the toolset encode-utils and its dependencies.
Next, you'll need to add your API access keys (these should have been provided to you by your data wrangler).
You can do this each session:
$ export DCC_API_KEY='xxxxxx' $ export DCC_SECRET_KEY='xxxxxxxx'
Or once globally:
-
Navigate to your
-
$ cd ~/ $ open .bash_profile
And add the lines:
-
export DCC_API_KEY='xxxxxx' export DCC_SECRET_KEY='xxxxxxxxx'
.bash_profile:
You may also specify environment variables corresponding to your lab and award objects' identifiers in the DCC_AWARD and DCC_LAB variables respectively in your .bash_profile, which will allow you to skip the award and lab fields when submitting object metadata with eu_register.py:
export DCC_AWARD='xxxxx' export DCC_LAB='your-lab-name'
The Submission example later in this document is written assuming that the DCC_AWARD and DCC_LAB environment variables have been defined. If you choose not to do this, be aware that lab and award are required properties you'll have to include in your tab-separated files for Biosample, Library, Experiment, File, and other objects.
Calling eu_register.py
encode_utils has a prepackaged script to allow you to submit to the Portal easily: eu_register.py. Proper installation should place the script in your /bin directory, so once you are in your python environment, you'll be able to run it from your terminal.
There are several arguments you'll need to include when using this script.
Required arguments:
-
mode
-mSpecification of the server you're submitting to. The ENCODE Portal (www.encodeproject.org) is referred to as the production server, and a special server mirroring production for testing purposes (www.test.encodedcc.org) is referred to as the test server.
The DCC strongly encourages you to start with submission to the test server (
-m dev), only proceeding to submission to the production server (-m prod) after validating correctness of the submission to the test server with your data wrangler's help. The two available modes are:-
-m dev(test.encodedcc.org) -
-m prod(encodeproject.org)
-
-
profile
-pSpecification of the object type you're submitting, for example:
-
-p biosample -
-p library -
-p genetic_modification
-
-
infile
-iSpecification of the tab-separated values file containing the metadata values for the object(s) being submitted. For example:
-i tab_separated_file.txt
Optional arguments:
-
Dry run
-dSpecification of a special mode of submission execution that will not result in actual object(s) creation on the Portal, but will validate to some extent the metadata properties specified in the tab-separated values file.
-
Submit object without aliases
--no-aliases:By default,
eu_register.pyrequires specification of aliases for any object that is being submitted. Use the flag--no-aliasesif you are interested in submitting objects without an alias. The use of this flag is discouraged by the DCC. - Help
-h
This option provides a summary of all the available arguments and flags to use with eu_register.py.
Log files with information relevant to your submissions will automatically be generated once you start to use the script.
Please continue to the Submission example to see how to format the tab_separated_file.txt (metadata) for several objects, and example commands for submission of these objects using eu_register.py.
Submission example
Biosample | Library | Experiment | Replicate | File
So, you're ready to submit the metadata for your experiment to the Portal. Here, we'll go through what the submission process should look like from start to finish for a new RNA-seq experiment on the cell line HepG2. This means we need to submit metadata for several distinct objects that constitute this experiment, and finally a file of results. The DCC recommends always starting with submission to the test environment (-m dev) before moving on to submission to the production server (-m prod).
For any object, you can always look at its schema profile to learn more about the various properties available.
Our RNA-seq example will be represented on the Portal with 5 types of elements (a.k.a. objects):
Biosample - representing the actual tube or sample of biological material used in the assay (here, a specific isolation/culture/sample of the cell line HepG2)
Library - representing the nucleic acid material derived from the biosample that will be used in the assay
Experiment - representing the assay performed on the biosample (RNA-seq in this case)
Replicate - representing the execution of the experiment to produce a library. (For example: if the assay was done on two distinct cell populations that grew independently, you would have two replicate objects, each with its own library and biosample.)
File - data file output of your experiment. These are usually raw data files generated by the specific assay. In our example we will submit a fastq file.
The order of submission matters. Since the objects are related (linked) to each other, creation of these relationships depends on the proper order of submission. For example, a library object is linked to a specific biosample object. Therefore, the biosample object needs to be submitted first.
Sometimes, you'll refer to preexisting objects on the Portal rather than making new ones. To avoid confusion, it's important not to have multiple objects on the Portal that refer to the same real-life entity. For example, if you're using a common cell line, the object representing the ontological term (BiosampleType.json) corresponding to the cell line should already be on the Portal. We'll cover how to look for preexisting objects (with BiosampleType as our example) at the end of the Biosample section.
Biosample
A culture of HepG2 cells, grown on a specific day, is a unique biosample. You may use this biosample to generate multiple replicates, each of which would refer to a different library prepared from the same biosample. Once you've submitted this particular biosample object, you'll be able to refer to it with its unique accession (ENCBS#) on the ENCODE Portal; if you have specified aliases upon submission you can use the alias you used to refer to the biosample object as well.
Some properties of your biosample, such as biosample_ontology, source, award, lab, and organism, you will describe by using an identifier for another object on the Portal! For example, when you specify "human" in the organism property of your biosample, that creates a link between your biosample object and an existing organism object named human. This allows you to sort and search biosamples by their linked donor objects.
Any object submitted to the Portal is validated against a JSON schema that describes the object being submitted. You will find the schemas for different objects here. Some of the schema properties are required upon submission (without these, the object will fail validation and you will see an error #422) and some of the properties are optional. The DCC strongly encourages you to submit rich metadata and include optional properties rather than submitting only the minimum required set.
The following properties are required for submission of biosamples:
-
biosample_ontology - a reference to an object representing an ontological description of your sample. Usually you can find the correct term by searching on the Portal (guidance at the end of this section). If you are unsure what BiosampleType you should use, please contact your data wrangler. Here, we'll refer to the existing BiosampleType for HepG2 cells by its alias, encode:HepG2.
-
organism - the species your biosample is derived from (human, mouse, dmelanogaster, or celegans)
-
source - the originating lab or vendor for your sample; a full list of sources registered on the Portal is available here. If you don't see your source, please contact your wrangler. Here, we're using /sources/atcc/
-
award - the award or grant this work was supported by
-
lab - the lab this work was performed in
In our example of a cell culture sample, we also recommend including the following:
-
culture_start_date - the day the culture was started (year-month-day)
-
culture_harvest_date - the day cells were harvested (year-month-day)
-
lot_id - the identifier for which lot of cells this sample originated from, via ATCC
-
product_id - the identifier for which cell product this is, via ATCC
-
starting_amount - the amount of sample you started with, an integer
-
starting_amount_units - the units describing the amount you started with
-
aliases - a lab-specific unique identifier starting with your lab name (john-doe:biosample_01)
Additional properties of the biosample object schema
For our example, the required data is collected in a table biosample.txt (always tab-separated):
| aliases | biosample_ontology | organism | source | culture_start_date | culture_harvest_date | lot_id | product_id | starting_amount | starting_amount_units |
| john-doe:biosample_01 | encode:HepG2 | human | /sources/atcc/ | 2019-01-02 | 2019-02-03 | 59635738 | HB-8065 | 3000 | cells |
Supply this to eu_register.py as follows:
$ eu_register.py -m dev -p biosample -i /path/to/biosample.txt
This command runs the submission script to submit a biosample object to the test server using metadata specified in biosample.txt. You will now be able to use the alias "john-doe:biosample_01" to refer to this biosample in other objects, but you can also use the accession your object was assigned upon submission. Once your object is successfully submitted (i.e., created), if you append its alias to the URL of the server you posted to, you'll be able to view your object. For our example: test.encodedcc.org/john-doe:biosample_01 will direct us to the new biosample object with a 'TSTBS' accession. This 'TSTBS' accession will be different from your final 'ENCBS' accession, which is assigned after submission to the production server.
Successful posting of your object (following the example above) will give you this output:
$ eu_register.py -m dev -p biosample -i biosample.txt
2019-12-16 12:43:17,525:eu_debug: Connecting to test.encodedcc.org
2019-12-16 12:43:17,526:eu_debug: submission=False: In non-submission mode.
2019-12-16 12:43:17,871:eu_debug: submission=True: In submission mode.
2019-12-16 12:43:17,876:eu_debug:
IN post().
2019-12-16 12:43:17,876:eu_debug: <<<<<< POST biosample record john-doe:biosample_01 To DCC with URL https://test.encodedcc.org/biosample and this payload:
{
"aliases": [
"john-doe:biosample_01"
],
"award": "U24HG009397",
"biosample_ontology": "encode:HepG2",
"culture_harvest_date": "2019-02-03",
"culture_start_date": "2019-01-02",
"lab": "/labs/john-doe/",
"lot_id": "59635738",
"organism": "human",
"product_id": "HB-8065",
"source": "/sources/atcc/",
"starting_amount": 3000,
"starting_amount_units": "cells"
}
2019-12-16 12:43:18,219:eu_debug: Success.
The script will catch object errors such as missing required properties, and fail to post. If you missed the source property in the example above, your output would include this error and explanation:
2019-12-16 12:36:15,367:eu_debug: Failed to POST john-doe:biosample_01
2019-12-16 12:36:15,367:eu_debug: <<<<<< DCC POST RESPONSE:
2019-12-16 12:36:15,367:eu_debug: {
"@type": [
"ValidationFailure",
"Error"
],
"code": 422,
"description": "Failed validation",
"errors": [
{
"description": "'source' is a required property",
"location": "body",
"name": []
}
],
"status": "error",
"title": "Unprocessable Entity"
}
The formatting of entries for each property also matters, and the script will throw an exception if you supply an entry that doesn't match what's expected. For more detail, see each object's schema.
Finding your BiosampleType on the Portal:
If you know the ontological term describing your sample (we use CL, EFO, and Uberon ontologies on the Portal), you can use this term to search the Portal for the predefined BiosampleType object you would use to specify your sample ontology.
For example: enter "HepG2" into the search bar, and select "BiosampleType" from the Data Type facet on the left:
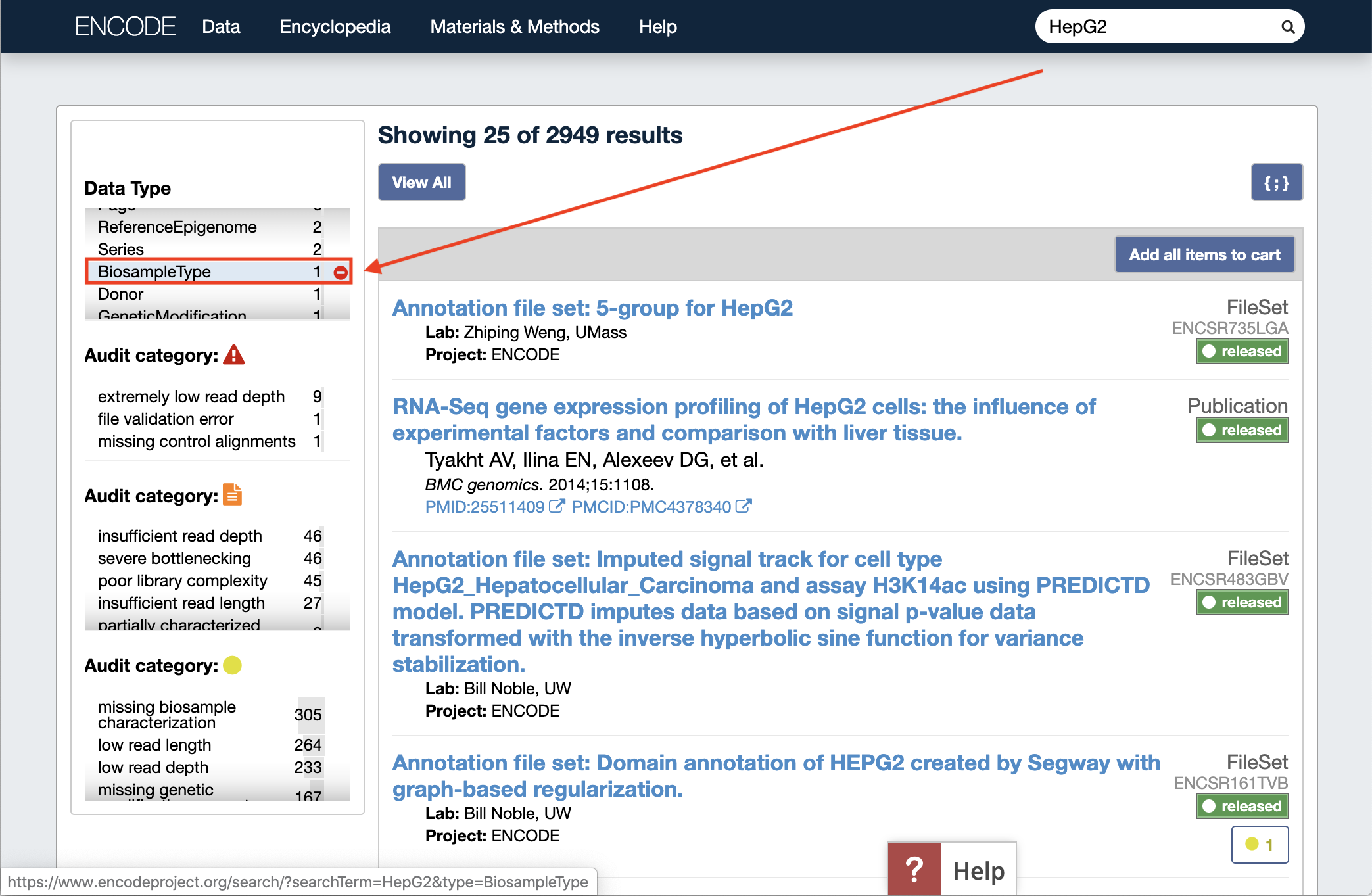
You will see this page of search results.
You can click the HepG2 (cell line) Biosample Type entry in the results, and then the {;} button on the upper right of the page to see the full JSON description for this BiosampleType object (we recommend using a JSON prettyprint plugin for readability in your browser. JSONView is available for Chrome and Firefox). This gives us identifiers for the BiosampleType for the example above that we would like to use, and we can specify HepG2 with the uuid given (ec2e6bce-118e-41e7-a1a3-f34f3278472c) or its alias (encode:HepG2).
You can also browse the existing list of BiosampleType objects by creating a search query for the object type, such as https://www.encodeproject.org/search/?type=BiosampleType. For any object type you'd like to browse existing entries for, you can append the schema or profile name to the Portal URL as follows: encodeproject.org/search/?type=ProfileName.
For more detailed help navigating and searching the Portal, you can read this. Please reach out to your data wrangler if you're not certain which BiosampleType to use.
Library
Using the biosample of HepG2 cells submitted above, you isolated total RNA to generate a library. This library object will help to link the correct files of experimental results with the nucleic acid material isolated from the biosample.
Library objects require these properties:
-
nucleic_acid_term_name - the material used (DNA, RNA, miRNA, protein, polyadenylated mRNA)
-
award - the award or grant this work was supported by
-
lab - the lab this work was performed in
We also recommend including these non-required properties to better describe our RNA-seq experiment:
-
biosample - the distinct accessioned sample this library is derived from (described in the previous section); you can refer to this with the accession (ENCBS#) or its alias (john-doe:biosample_01)
-
extraction_method - the method used to extract the nucleic acids (such as: Ambion mirVana, Trizol, or Qiagen DNeasy Blood & Tissue Kit; enum listed here in Properties:extraction_method). If you don't see your specific method listed, reach out to your data wrangler to have it added.
-
rna_integrity_number - an assessment of RNA quality (RIN value)
-
depleted_in_term_name - the specific subset of nucleic acid diminished from the library (capped mRNA, polyadenylated mRNA, or rRNA)
-
aliases - a lab-specific unique identifier starting with your lab name (john-doe:library_01)
Additional properties of the library object schema
For our example, the metadata in tab-separated format becomes file library.txt:
| aliases | nucleic_acid_term_name | biosample | extraction_method | rna_integrity_number | depleted_in_term_name |
| john-doe:library_01 | RNA | john-doe:biosample_01 | Qiagen RNA extraction | 8.7 | rRNA |
Supply this to eu_register.py as follows:
$ eu_register.py -m dev -p library -i /path/to/library.txt
This command runs the submission script to submit a library object to the test server using metadata specified by library.txt. Notice that a biosample identifier (alias) is included- this means that the metadata object representing the biosample that this library was prepared from should be submitted first in order to use its accession. Remember that you can also use an accession to specify a biosample object.
Experiment
In order to describe the assay being performed, you will create an experiment object. This also provides a way to connect the Biosample(s), Library(-ies), resulting File(s), and other details from an assay in our data model to be visualized as a single page on the Portal.
Experiment objects require the following properties:
-
assay_term_name - the name of the assay (such as: ChIP-seq, long read RNA-seq, or eCLIP; enum listed here under Properties:assay_term_name). In this example, RNA-seq
-
biosample_ontology - this should match the ontology specified in your biosample (described above)
-
award - the award or grant this work was supported by
-
lab - the lab this work was performed in
While not required, it is helpful to include:
-
description - a short, plain text description of your experiment
-
aliases - a lab-specific unique name starting with your lab name (john-doe:experiment_01)
Additional properties of the experiment object schema
For our example, the required data in tab-separated format becomes file experiment.txt:
| aliases | assay_term_name | biosample_ontology | description |
| john-doe:experiment_01 | RNA-seq | encode:HepG2 | My best RNA-seq experiment yet |
Supply this to eu_register.py as follows:
$ eu_register.py -m dev -p experiment -i /path/to/experiment.txt
Replicate
Using the library of total RNA from the biosample of HepG2 cells, RNA-seq is performed. A replicate object is a mapping between the experiment and a library, so each replicate that is part of an overall experiment references a unique library.
Replicate objects require the following properties:
-
experiment - the experiment the replicate is part of (described above)
-
biological_replicate_number - one per biosample; in this case, our replicate is biological_replicate_number:1
-
technical_replicate_number - may be multiple per biosample; in this case, our replicate is technical_replicate_number:1
We also strongly recommend including:
-
library - the library being sequenced or assayed (described above)
-
aliases - a lab-specific unique identifier starting with your lab name (john-doe:replicate_01)
Additional properties of the replicate object schema
For our example, the required data in tab-separated format becomes file replicate.txt:
| aliases | experiment | biological_replicate_number | technical_replicate_number | library |
| john-doe:replicate_01 | john-doe:experiment_01 | 1 | 1 | john-doe:library_01 |
Supply this to eu_register.py as follows:
$ eu_register.py -m dev -p replicate -i /path/to/replicate.txt
This command runs the submission script to submit a replicate object to the test server using metadata specified by replicate.txt. Note that we used an alias to specify both our experiment and library objects, but the ENC accessions and the aliases are each valid to refer to a submitted object.
File
After running an RNA-seq experiment as described with our objects above, there's a resulting fastq file to submit.
For files, the following fields are required:
-
dataset - the dataset or experiment accession (or alias) the file will be associated with
-
file_format - the format of the file, such as bam, bed, or vcf. The list of valid enums is here under Properties:file_format
-
output_type - a description of the file's purpose or contents, such as: chromatin interactions, filtered peaks, or variant calls. The list of valid enums is here under Properties:output_type
-
md5sum - a calculated digital fingerprint of your file, which will be unique to the file you are going to submit. While required, you won't need to specify it in your
input_file.txtaseu_register.pywill calculate it for you. -
submitted_file_name - the name of and path to your file locally. This will allow the upload process (see next step) to take place automatically.
-
award - the award or grant this work was supported by
-
lab - the lab this work was performed in
Additional required properties will vary based on your file_format, and the schema for File objects is available here for specific details. For fastq files, you will need these additional properties:
-
replicate - the uuid (or alias) for the replicate the fastq file is from (described above)
-
run_type - single-ended or paired-ended
-
read_length - an integer for the length of the sequencing reads
-
platform - the machine used for generating sequence data, such as: Illumina NovaSeq 6000, Illumina NextSeq 500, or Pacific Biosciences Sequel II. The complete list of options is available here
We also strongly recommend including:
-
aliases - a lab-specific unique identifier starting with your lab name (john-doe:fastqfile_01)
Additional properties of the file object schema
For our example, the required data in tab-separated format becomes file input_file.txt:
| dataset | file_format | run_type | submitted_file_name | platform | output_type | replicate | read_length | aliases |
| ENCSR### | fastq | single-ended | /pathto/file.fastq.gz | encode:MiSeq | reads | uuid### | 50 | john-doe:fastqfile_01 |
Now that you have your metadata in order, you can use the eu_register.py script to submit your file:
$ eu_register.py -m dev -p file -i /path/to/input_file.txt
This command will run the submission script to submit a file object to the test server using metadata from input_file.txt.
Special note:
If your run_type is paired-ended, you must also include paired_with - an accession (or alias) of the file the fastq is paired with. This should be specified for read2 files only; the read1 paired_with property is calculated.
Uploading step
Specifically, when submitting file object metadata, there is an extra step that uploads the actual data file you're describing to an S3 bucket associated with ENCODE.
If the file is on your computer and you included submitted_file_name in your metadata, this upload happens automatically when you run the eu_register.py command for a file object given above. If your file is in an S3 bucket, you can follow the instructions here: https://github.com/StanfordBioinformatics/encode_utils/wiki/could-to-cloud-file-transfers#s3-to-s3 to transfer from S3 to S3 directly.
Success!
You've submitted the metadata and files for your experiment!
Further reading
The official documentation for encode_utils is available at Read the Docs, including specific guidance for eu_register.py.
Getting Started - Using the Portal
Data Organization - Data Model, Accessions, Ontologies
If you need additional help, please reach out to your data wrangler. You can also contact the ENCODE help-desk at encode-help@lists.stanford.edu