Small RNA-seq Data Standards and Processing Pipeline
Assay Overview
There are many classes of small RNAs (tRNAs, microRNAs, miRNA, snoRNAs, etc…) less than 200 nucleotides in length, each of which carries out distinct biochemical roles in the cell. These roles may include messenger RNA interference silencing and degredation, among others. These RNAs can be captured and sequenced using a variety of methods to enrich for various classes.
Updated June 2017
Pipeline Overview
The small RNA-seq pipeline was developed as a part of the ENCODE Uniform Processing Pipelines series. The full pipeline code is freely available on Github and can be run on DNAnexus (link requires account creation) at their current pricing.
The ENCODE RNA-seq pipeline for small RNAs can be used for libraries generated from rRNA-depleted total RNA that are size-selected to be shorter than approximately 200 nucleotides. Data may be paired-ended and stranded or single-ended and unstranded.
Pipeline Schematic
View the current instances of this pipeline for single-ended experiments
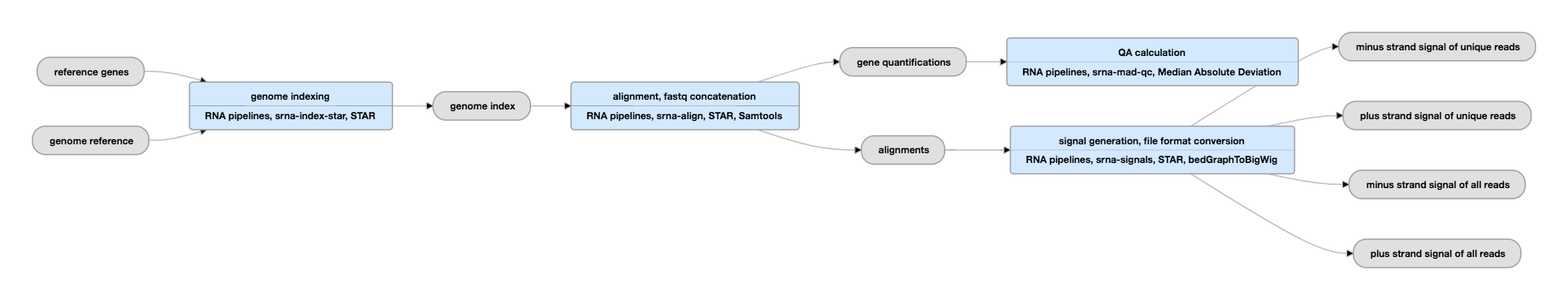
Inputs:
| File format |
Information contained in file |
File description |
Notes |
| fastq | reads | Single-ended, stranded, g-zipped small RNA-seq reads | Reads must meet the criteria outlined in the Uniform Processing Pipeline Restrictions. |
| gtf | genome annotation | Default genome annotation file is from GENCODE. | Human experiments use GENCODE V24 or V19, while mouse use GENCODE M4. |
Outputs:
| File format | Information contained in file |
File description |
Notes |
| bam |
alignments |
Produced by mapping reads to the genome. | |
| bigWig | signal | Normalized RNA-seq signal | Signals are generated for unique reads and unique+multimapping reads in both the plus and minus strands. |
| tsv | gene quantifications | STAR-generated outputs | The four columns of the file are as follows:
|
| The pipeline also produces quality metrics, including gene quantification level and read depth. | |||
References
Genomic References
View the mapping assembly references and STAR genome index used in this pipeline
Links and Publications
Find data generated by this pipeline here
Explore publications (in progress)
Uniform Processing Pipeline Restrictions
- The read length should be a minimum of 50 base pairs.
- Sequencing should be single-ended.
- All Illumina platforms are supported for use in the uniform pipeline; colorspace (SOLiD) are not supported.
- Barcodes and spike-in sequences, if present in the fastq, must be indicated in the flowcell metadata.
- Library insert size range must be indicated.
- Alignment files are mapped to either the GRCh38, hg19, or mm10 sequences.
- Gene and transcript quantification files are annotated to either GENCODE V24, V19, or M4.
Current Standards
Experimental guidelines for small RNA-seq experiments can be found here.
- An small RNA-seq experiment is an RNA-seq assay in which the average library insert size is <200 base pairs.
- Experiments should have two or more replicates. Assays performed using EN-TEx samples may be exempted due to limited availability of experimental material.
- Each replicate should have 30 million aligned reads, although older projects aimed for 10 million aligned reads. Best practices for ENCODE2 RNA-seq experiments have been outlined here.
- Replicate concordance:the gene level quantification should have a Spearman correlation of >0.9 between isogenic replicates and >0.8 between anisogenic replicates (i.e. replicates from different donors).
- The experiment must pass routine metadata audits in order to be released.