Bulk RNA-seq Data Standards and Processing Pipeline
Assay overview
RNA-seq measures the quantity and sequences of ribonucleic acid material. Bulk RNA-seq experiments are specifically designed to gather information on RNA libraries where the average insert size is greater than 200 bases.
Updated May 2021
Pipeline overview
The ENCODE4 version of the bulk RNA-seq pipeline was developed as a part of the ENCODE Uniform Processing Pipelines series. All code for this pipeline is freely available on Github.
The ENCODE Bulk RNA-seq pipeline can be used for both replicated and unreplicated, paired-ended or single-ended, and strand-specific or non-strand specific RNA-seq libraries. Libraries must be generated from mRNA (poly(A)+, rRNA-depleted total RNA, or poly(A)-) populations that are size-selected to be longer than approximately 200 bp. The pipeline performs alignment, generates signal tracks, and quantifies genes and isoforms.
Pipeline schematic
View the current instance of this pipeline
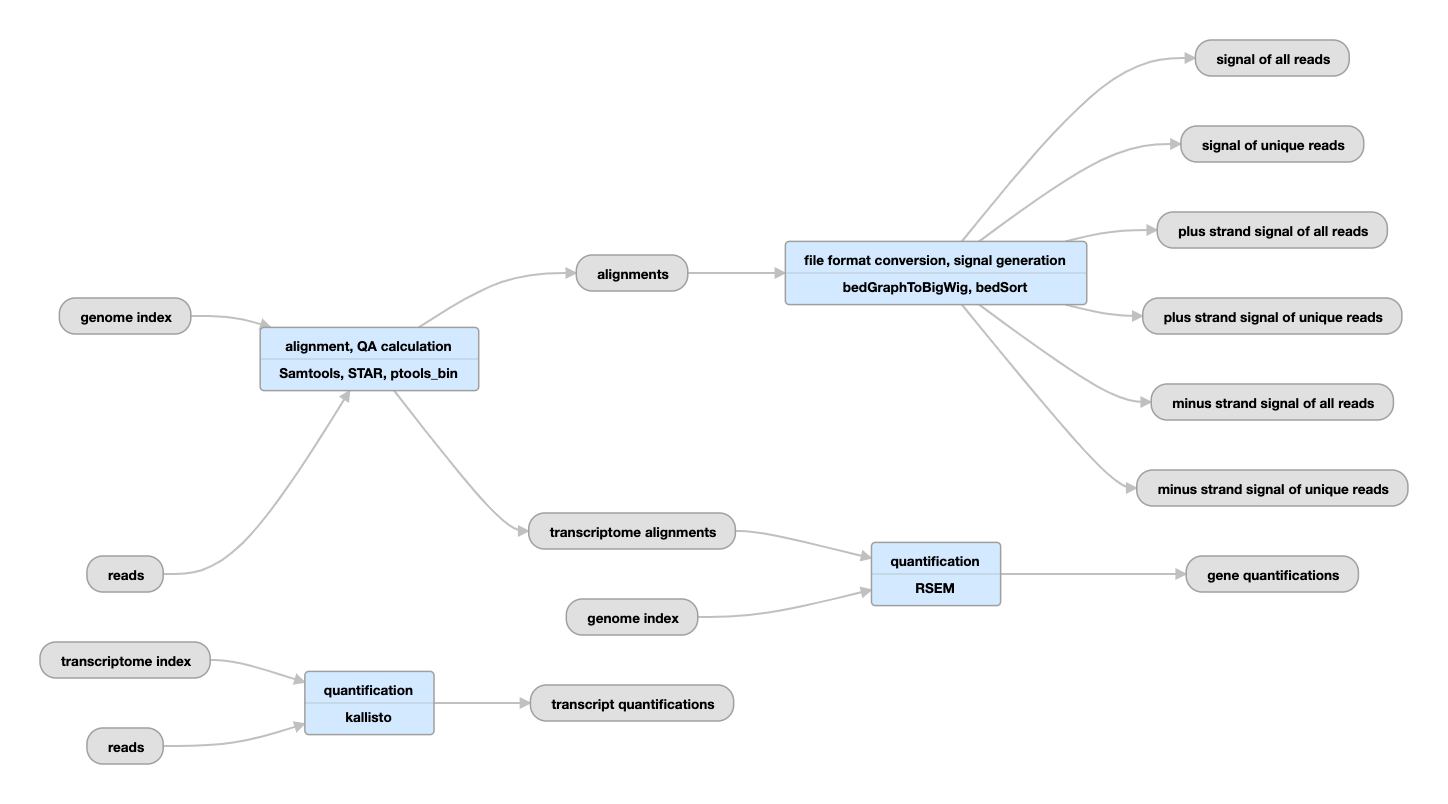
Inputs:
| File format |
Information contained in file |
File description |
Notes |
| fastq |
reads |
G-zipped bulk RNA-seq reads | Reads must meet the criteria outlined in the Uniform Processing Pipeline Restrictions. |
| tar | genome index | Generated by STAR or TopHat | Please see the paragraph titled "Regarding alignment and quantification" below the "Outputs" table for more on the aligners and their indices. |
| tar | transcriptome index | Index generated by kallisto | |
| fasta | spike-ins | ERCC Spike-ins (External RNA Control Consortium) | The spike-in sequences are effectively the controls for the RNA-seq experiment. |
Outputs:
| File format |
Information contained in file |
File description |
Notes |
| bam | alignments | Produced by mapping reads to the genome. | Please see the paragraph titled "Regarding alignment and quantification" below the "Outputs" table for more on the aligners and their indices. |
| bam | transcriptome alignments | Produced by mapping reads to the transcriptome. | |
| bigWig | signal | Normalized RNA-seq signal | For stranded data, signals are generated for unique reads and unique+multimapping reads in both the plus and minus strands. For unstranded data, signals are generated for unique reads and unique+multimapping reads without regard for strand identity. |
| tsv | gene quantifications | Quantifications of reads (or read pairs, in paired-end sequencing) aligning to the gene annotation reference |
The file format specifications are as follows:
|
| tsv | transcript quantifications | Please see the caution regarding transcript quantifications in the paragraph below titled "Regarding alignment and quantification". | |
| The pipeline also produces quality metrics, including Spearman correlation and read depth. | |||
Regarding alignment and quantification:
The mapping of the reads is done using the STAR program, the quantification of transcripts is done with kallisto, and gene quantifications are generated using the RSEM program. Although there is general agreement between the mappings and the gene quantifications produced by different RNA-seq pipelines, quantifications of individual transcript isoforms, being much more complex, can differ substantially depending on the processing pipeline employed and are of unknown accuracy. Therefore, alignments and gene quantifications can be used confidently, while transcript quantifications should be used with care.
References
Genomic References
View the genome references and chromosome sizes used in this pipeline in human and mouse.
These pipelines require both assembly information for the species of interest and gene and transcript references. Each of the programs RSEM, kallisto, and STAR create indices for use in subsequent steps. More information on the use of RSEM is available here.
Exogenous RNA spike-in controls
Exogeneous RNA spike-in controls are added to samples to create a standard baseline for the quantification of RNA expression (PMC3166838). The ENCODE consortium is standardizing on the use of the Ambion Mix 1 commercially available spike-ins at a dilution of ~2% of final mapped reads. However, there is a mixture of older data and imported data. Therefore, to track the spike-ins used in a given library, there is a dataset associated with the library. That dataset will contain the spike-ins sequence file in fasta format and information on the concentrations. These spike-in sequences are expected to be found in the genome index used in the mapping step(s) and in the subsequently generated bam. The quantifications of the sequences can be found in the RSEM transcript and gene quantification files.
View spike-ins datasets
View the certificate of analysis for ERCC spike-ins
Access the ERCC dash board
Links and Publications
Find data generated by this pipeline: All | paired-end only | single-end only
Uniform Processing Pipeline Restrictions
- The read length should be a minimum of 50 base pairs.
- Sequencing may be paired- or single-end, as long as sequencing type is specified and read pairs are indicated.
- All Illumina platforms are supported for use in the uniform pipeline; colorspace (SOLiD) are not supported.
- Barcodes, if present in fastq, must be indicated in the flowcell metadata.
- ERCC spike-ins should be used in library preparation with the concentrations indicated in the metadata.
- Library insert size range must be indicated. For kallisto quantification in single-end libraries, the average fragment size and SD or CV of the sizes must be provided.
- Alignment files are mapped to the GRCh38 or mm10 sequences.
- Gene and transcript quantification files are annotated to GENCODE V29 or M21.
Current Standards
Experimental guidelines for bulk RNA-seq experiments can be found here.
- A bulk RNA-seq experiment is an RNA-seq assay in which the average library insert size is 200 base pairs.
- Experiments should have two or more replicates. Assays performed using EN-TEx samples may be exempted due to limited availability of experimental material.
- Each replicate should have 30 million aligned reads, although older projects aimed for 20 million reads. Best practices for ENCODE2 RNA-seq experiments have been outlined .
- Replicate concordance: the gene level quantification should have a Spearman correlation of >0.9 between isogenic replicates and >0.8 between anisogenic replicates (i.e. replicates from different donors).
- The experiment must pass routine metadata audits in order to be released.
Single-cell Isolation followed by RNA-seq Specific Standards
- Experiments are in sets of 10 to 20 individual experiments, which are not considered biologically replicated.
- Each replicate requires only 5 million aligned reads.
- Each experiment should have a corresponding cell-equivalent control experiment.
shRNA Knockdown Followed by RNA-seq and CRISPR Genome Editing Followed by RNA-seq Specific Standards
- Each replicate should have 10 million aligned reads.
- The target of the knockdown must be defined.
- Each experiment should have a corresponding control experiment.
siRNA knockdown
- Each replicate should have 10 million aligned reads.
- Each experiment should have a corresponding control experiment.
- Replicate concordance: the gene level quantification should have a Spearman correlation of >0.9 between isogenic replicates and >0.8 between anisogenic replicates.
- % knockdown of the targeted factor should be verified for each replicate relative to the control.