DNase-seq Data Standards and Processing Pipeline
Assay overview
DNaseI-seq, or DNase-seq as it is referred to on the ENCODE portal, is a global and high-resolution method that uses the non-specific endonuclease DNaseI to map chromatin accessibility. These accessible regions, designated as DNaseI hypersensitive sites (DHSs), define the regulatory features, (eg. promoters, enhancers, insulators, locus control regions) of complex genomes. DNase-seq is an unbiased and robust method that is not predicated on an a priori understanding of regulatory patterns or chromatin features.
Updated Dec 2020
Pipeline Overview
The ENCODE 4 DNase-seq pipeline was developed in collaboration with the Stamatoyannopoulos lab as a part of the ENCODE Uniform Processing Pipelines series. The pipeline for processing DNase I sequencing data takes in Illumina sequencing reads from DNase I experiments, assess the data, and returns genomic regions with statistically significant enrichments, or 'hotspots', of cleavage activity from the original DNase I experiment. The full DNase-seq pipeline code is available on Github.
DNase-seq pipeline
View the current pipeline instance.
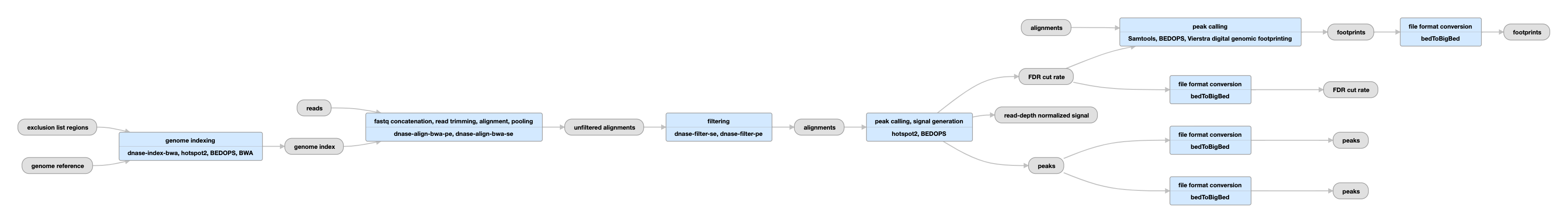
Inputs:
| File format |
Information contained in file |
File description |
Notes |
| fastq |
reads |
G-zipped reads, paired-ended or single-ended. | Multiple FASTQs from a single biological replicate or library are concatenated before mapping |
| fasta | genome indices, genome reference | Indices are dependent on the assembly being used for mapping | |
| txt |
chromosomes reference |
List of expected chromosomes | |
| bed, starch | hotspots reference | Reference files used for hotspots and peaks calling | Hotspots reference must be selected guided by the input FASTQ read length |
| txt | bias model | Reference file used for footprints modeling |
Outputs:
| File format |
Information contained in file |
File description |
Notes |
| bam | unfiltered alignments | FASTQs are concatenated, trimmed for adapters and length, and aligned with BWA | |
| bam | alignments | unfiltered alignments are filtered and deduplicated | |
| bigWig | read-depth normalized signal | Density starch from Hotspot2 is normalized and converted to a bigWig | |
| bed and bigBed narrowPeak | peaks | DNase I hypersensitivity sites identified using Hotspot2 | Both 5% and 0.1% FDR peak calls are included in the outputs. FDR stands for False Discovery Rate. |
| bed and bigBed bed3+ |
FDR cut rate |
FDR estimate per site for being called hypersensitive to cleavage by DNase I | |
| bed and bigBed bed3+ |
footprints |
A footprint model is fit and statistical deviations from expected cleavage rates within hotspot regions are called and thresholded |
1% FDR footprints. A detailed explanation of the footprint modeling is available in the footprint-tools documentation. |
| starch |
peaks |
DNase I hypersensitivity sites identified using Hotspot2 | 0.1% FDR peaks |
References
Genomic References
View the reference files used in this pipeline
Links and Publications
Find data generated by the ENCODE 4 DNase-seq pipeline
Back to the top
Current Standards
Experimental guidelines for DNase experiments can be found here.
- Experiments should have two or more biological replicates, isogenic or anisogenic. Assays performed using EN-TEx samples or other rare types may be exempted due to limited availability of experimental material.
- A SPOT (Signal Portion of Tags) score of 0.4 or higher is considered a product of high quality data.
- A SPOT score of 0.25 is considered minimally acceptable for rare and hard to find primary tissues. In very rare cases of limited sample availability, lower scoring data may be used with appropriate caution.
- Any sample with a SPOT score <0.3 should be targeted for replacement with a higher quality sample, whenever possible.
- SPOT scores should be calculated on de-duplicated data or on data for which the duplicate rates are <5%.
- DNase-seq requires a minimum of 20 million uniquely mapping reads to generate a reliable SPOT score, and 100 million uniquely mapping reads to generate reliable DNase footprints.
- For a standard conventional DNase-seq profile, 50 million uniquely mapping reads are recommended.
- For deep, footprinting depth DNase-seq, a depth of 150-200 million uniquely mapping paired-end reads are recommended.
- Acceptable mappability rates and mitochondrial content are listed below:
| SPOT Score* | Mitochondrial Fraction | Mappability (without mitochondrial fraction) |
Resulting Data Status |
|---|---|---|---|
| ≥ 0.4 | < 10% | > 75% | Good |
| 0.3 - 0.4 | 10 - 15% | 65 - 75% | Acceptable |
| < 0.25 | > 15% | < 65% | Poor |
| * SPOT scores of 0.25 - 0.3 are minimally acceptable in certain circumstances (see above explanation) | |||
- Replicate concordance: the gene level quantification should have a Pearson correlation of >0.9 between isogenic replicates and >0.85 between anisogenic replicates.
- The experiment must pass routine metadata audits in order to be released.
Uniform Processing Pipeline Restrictions
- The read length should be a minimum of 36 base pairs.
- Read trimming of adapter sequences is recommended.
- Failure to trim sequences of fragments sized shorter than the sequencing cycle number can lead to filtering small fragment signal and create bias in the resulting alignments.
- There is no post-trimming read length minimum, but effective fragment mapping drops significantly at kmer-lengths of less than 22 base pairs.
- Adapter sequences used in library creation should be documented and available to the pipeline.
- Barcodes/UMI coding should be indicated in the metadata and available for accurate application of duplication filtering methods by the pipeline.
- Sequencing may be paired- or single-end, as long as sequencing type is specified and read pairs are indicated. Paired-end sequencing is preferred.
- The sequencing platform used must be indicated.
- Alignment files are mapped to either the GRCh38 or mm10 sequences.