ENCODE3 Transcription Factor ChIP-seq Data Standards and Processing Pipeline
Assay overview
ChIP-seq is a method used to analyze protein interactions with DNA. ChIP-seq combines chromatin immunoprecipitation with DNA sequencing to infer the possible binding sites of DNA-associated proteins. The ENCODE consortium has developed two analysis pipelines to study two different classes of protein-chromatin interactions. The transcription factor ChIP-seq (TF ChIP-seq) pipeline, described here, is suitable for proteins that are expected to bind in a punctate manner, such as to specific DNA sequences or specific chromatin configurations. In these assays, the IP target is typically a known or putative transcription factor or chromatin remodeler but can also be an RNA-binding protein or other DNA- or chromatin-specific factor. The pipeline for histone ChIP-seq is available here, and is best for proteins that associate with DNA over longer regions or domains.
Updated July 2020
Pipeline Overview
The ChIP-seq transcription factor pipeline was developed as a part of the ENCODE Uniform Processing Pipelines series. The ENCODE Consortium has developed two analysis pipelines to study the different classes of protein-chromatin interactions. Both ChIP-seq pipelines share the same mapping steps, but differ in the methods for signal and peak calling and in the subsequent statistical treatment of replicates. The full ChIP-seq pipeline code is available on Github.
Transcription factor ChIP-seq (TF ChIP-seq) specifically looks at proteins which are thought to associate with specific DNA sequences to influence the rate of transcription.
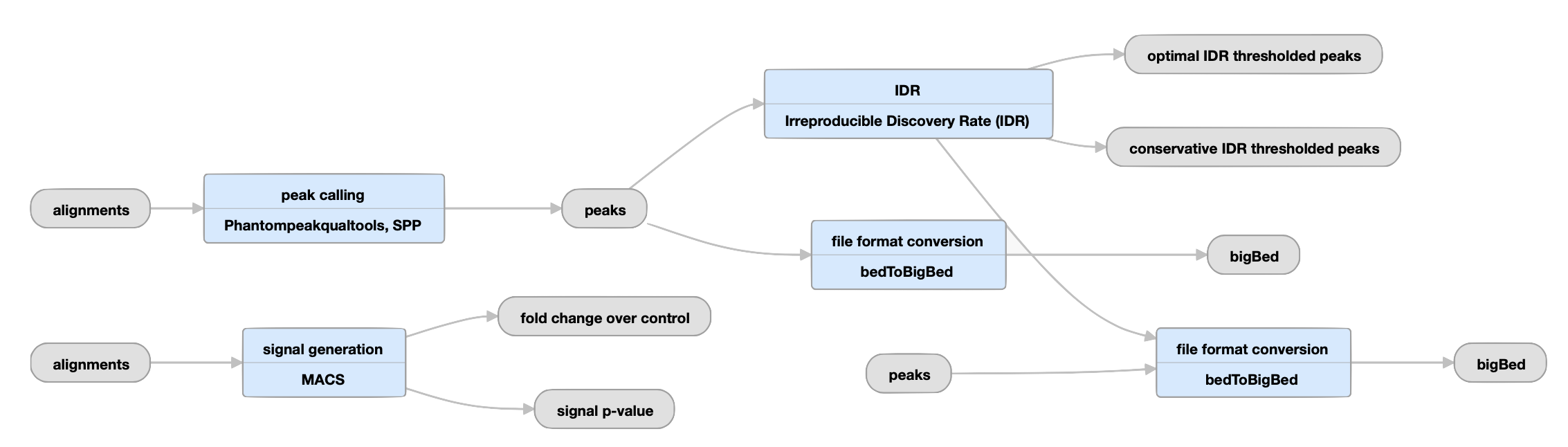
Replicated experiments
View the ENCODE3 replicated TF pipeline instance.
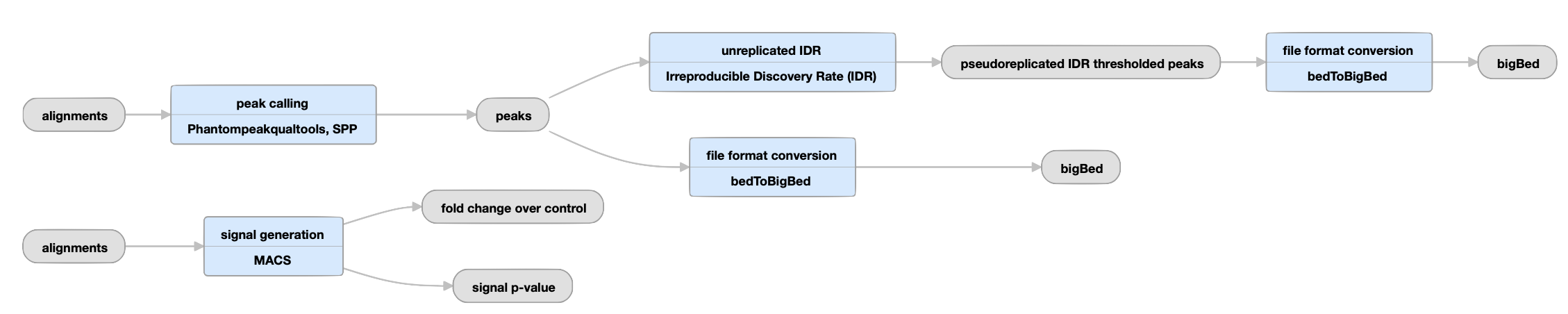
Unreplicated experiments
View the ENCODE3 unreplicated TF pipeline instance.
Back to the top
Inputs:
| File format |
Information contained in file |
File description |
Notes |
| fastq |
reads |
G-zipped reads, paired-ended or single ended, stranded or unstranded | Multiple fastqs from a single biological replicate or library are concatenated before mapping. Reads must meet the criteria outlined under the Uniform Processing Pipeline Restrictions. |
| fasta | genome indices | Indices are dependent on the assembly being used for mapping |
Outputs:
| File format |
Information contained in file |
File description |
Notes |
| bigWig | fold change over control, signal p-value | Two versions of nucleotide resolution signal coverage tracks. |
The signal is expressed in two ways: as fold-over control at each position, and as a p-value to reject the null hypothesis that the signal at that location is present in the control. |
|
bed and bigBed (narrowPeak) |
peaks |
Relaxed peak calls for each replicate individually and for both replicates reads pooled together. |
These peaks are thresholded to sample enough noise in the experiment for efficient statistical comparison of replicates in subsequent steps; as such, many false positives are expected to be present. They are not meant to be interpreted as definitive binding events, but are rather intended to be used as input for subsequent statistical comparison of replicates. |
|
|
|||
| bed and bigBed (narrowPeak) | conservative IDR peaks |
Peaks derived from IDR analysis of biological replicates. |
IDR, or Irreproducible Discovery Rate, measures the reproducibility of the assay in question. The optimal set of peaks is more sensitive, especially when one replicate has substantially lower data quality than the other. |
| bed and bigBed (narrowPeak) | optimal IDR peaks | The largest set of peaks derived from IDR analysis of biological replicates and pseudoreplicates. | |
|
|
|||
| bed and bigBed (narrowPeak) | pseudoreplicated IDR thresholded peaks | The set of peak calls from two partitions, or "pseudoreplicates" |
In the "unreplicated IDR" step, an IDR-based strategy is used to identify stable peaks across pseudoreplicates (instead of across true replicates). Peaks from the relaxed set (from all reads) as well as from pseudoreplicates are ranked, and those peaks that are well-supported in both pseudoreplicates (i.e. cross the same IDR threshold as for replicated experiments) are reported. There are no corresponding optimal and conservative peak sets in the unreplicated case because there are no true replicates. |
|
Quality control metrics are collected to determine library complexity, read depth, FRiP score, and reproducibility. |
|||
References
Genomic References
View the human (GRCh38) and mouse (mm10) reference files used in this pipeline
Links and Publications
Find data generated by the transcription factor pipeline for replicated experiments
Find data generated by the transcription factor pipeline for unreplicated experiments
Explore all ChIP-seq related publications on the ENCODE portal
Back to the top
Uniform Processing Pipeline Guidelines
-
The read length should be a minimum of 50 base pairs, though longer read lengths are encouraged; the pipeline can process read lengths as low as 25 base pairs. Sequencing may be paired- or single-ended.
- The sequencing platform used should be indicated.Different sequencing platforms might not be comparable. For example, replicates of HiSeq2000 vs. HiSeq4000 are different, and not comparable.
- Replicates should match in terms of read length and run type.
- Pipeline files are mapped to either the human (GRCh38) and mouse (mm10) sequences.
Back to the top
ENCODE3 Standards
*Note: Current ENCODE4 standards are found here.
Experimental guidelines for ENCODE3 ChIP-seq and epitope-tagged ChIP-seq experiments can be found here.
- Experiments should have two or more biological replicates, isogenic or anisogenic. Assays performed using EN-TEx samples may be exempted due to limited availability of experimental material.
- Antibodies must be characterized according to standards set by the ENCODE Consortium. Please see the linked documents for transcription factor standards (May 2016) and Epitope-tagged Transcription Factor standards (August 2019).
- Each ChIP-seq experiment should have a corresponding input control experiment with matching run type, read length, and replicate structure.
- Library complexity is measured using the Non-Redundant Fraction (NRF) and PCR Bottlenecking Coefficients 1 and 2, or PBC1 and PBC2. Preferred values are as follows: NRF>0.9, PBC1>0.9, and PBC2>10.
- The experiment must pass routine metadata audits in order to be released.
Target-specific Standards
- Each replicate should have 20 million usable fragments.
- low read depth: 10 million to 20 million usable fragments
- insufficient read depth: 5 million to 10 million usable fragments
- extremely low read depth: < 5 million usable fragments
- For transcription factor experiments, replicate concordance is measured by calculating IDR values (Irreproducible Discovery Rate). The experiment passes if both rescue and self consistency ratios are less than 2.
Additional Metrics
- Additional metrics are calculated without defined thresholds, such as fraction of reads in peaks (FRiP), and are useful in comparing similar experiments. These metrics are described here.
ENCODE2 Standards (Previous Standards)
Data quality standards for ENCODE2 are outlined in ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia.
- Experiments should have two or more biological replicates, isogenic or anisogenic.
- Antibodies must be characterized according to standards set by the consortium for ENCODE2 data.
- Each ChIP-seq experiment should have a corresponding input or IgG control experiment with matching run type, read length, and replicate structure.
- Library complexity is measured using the Non-Redundant Fraction (NRF) and PCR Bottlenecking Coefficients 1 and 2, or PBC1 and PBC2. Preferred values are as follows: NRF>0.9, PBC1>0.9, and PBC2>10.
- The experiment should pass routine metadata audits in order to be released.
Target-specific Standards
- Each replicate should have 10 million usable fragments.
- For transcription factor experiments, replicate concordance is measured by calculating IDR values (Irreproducible Discovery Rate). The experiment passes if both rescue and self consistency ratios are less than 2.